Division of Biostatistics, University of California, Berkeley, Berkeley, CA, USA.
BMC Bioinformatics. 2010 Feb 18;11:94. doi: 10.1186/1471-2105-11-94.
High-throughput sequencing technologies, such as the Illumina Genome Analyzer, are powerful new tools for investigating a wide range of biological and medical questions. Statistical and computational methods are key for drawing meaningful and accurate conclusions from the massive and complex datasets generated by the sequencers. We provide a detailed evaluation of statistical methods for normalization and differential expression (DE) analysis of Illumina transcriptome sequencing (mRNA-Seq) data.
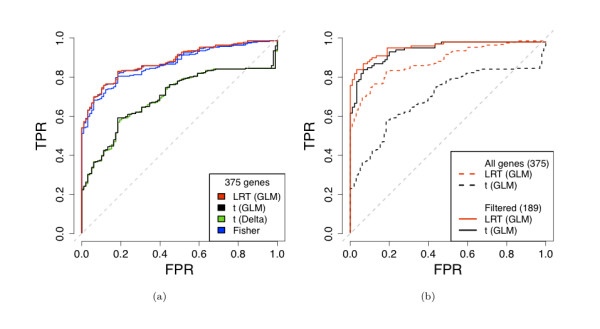
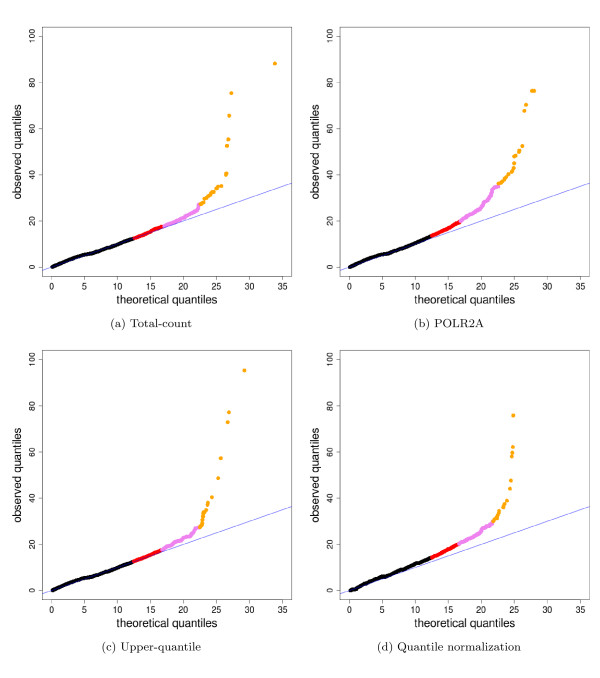
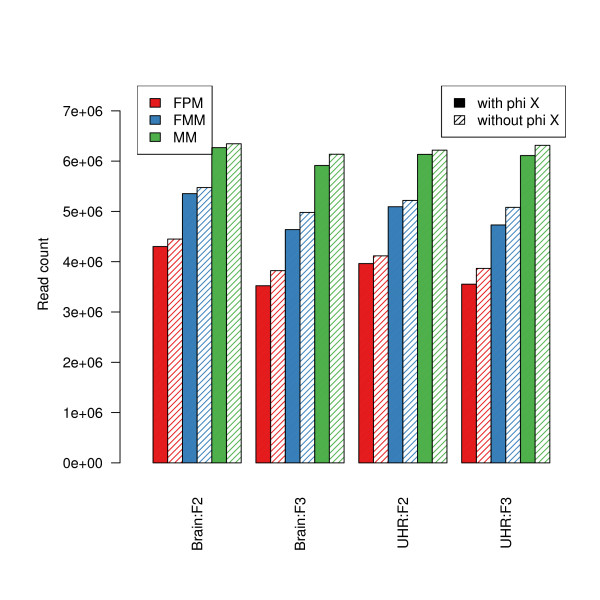
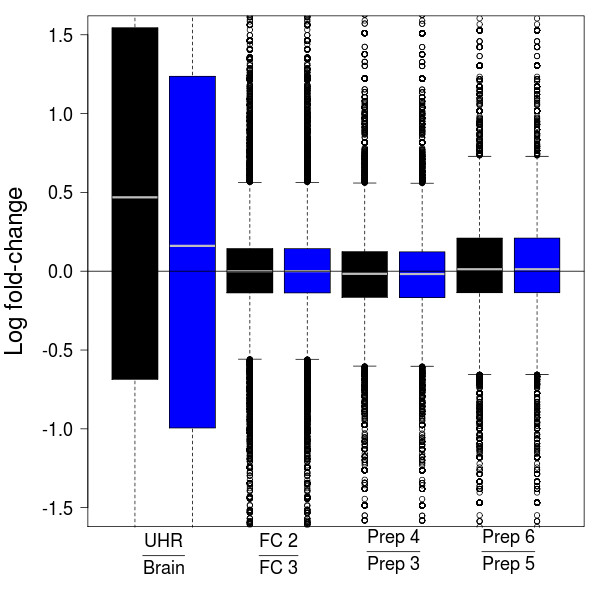
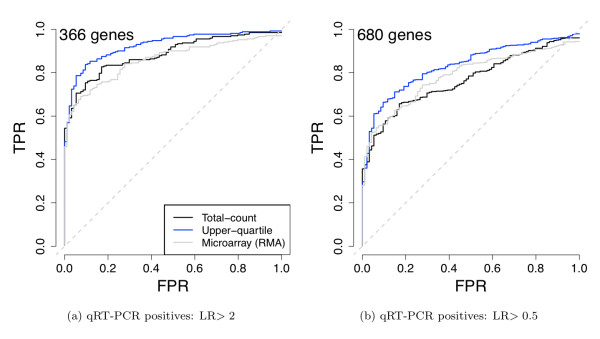
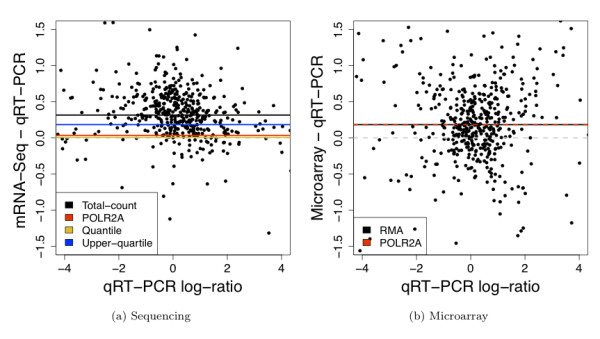
We compare statistical methods for detecting genes that are significantly DE between two types of biological samples and find that there are substantial differences in how the test statistics handle low-count genes. We evaluate how DE results are affected by features of the sequencing platform, such as, varying gene lengths, base-calling calibration method (with and without phi X control lane), and flow-cell/library preparation effects. We investigate the impact of the read count normalization method on DE results and show that the standard approach of scaling by total lane counts (e.g., RPKM) can bias estimates of DE. We propose more general quantile-based normalization procedures and demonstrate an improvement in DE detection.
Our results have significant practical and methodological implications for the design and analysis of mRNA-Seq experiments. They highlight the importance of appropriate statistical methods for normalization and DE inference, to account for features of the sequencing platform that could impact the accuracy of results. They also reveal the need for further research in the development of statistical and computational methods for mRNA-Seq.
高通量测序技术,如 Illumina 基因组分析仪,是研究广泛的生物和医学问题的强大新工具。统计和计算方法是从测序仪生成的大量复杂数据集得出有意义和准确结论的关键。我们提供了对 Illumina 转录组测序 (mRNA-Seq) 数据进行标准化和差异表达 (DE) 分析的统计方法的详细评估。
我们比较了用于检测两种类型的生物样本之间差异表达基因的统计方法,发现测试统计数据在处理低计数基因方面存在很大差异。我们评估了测序平台的特征,例如基因长度变化、碱基调用校准方法(带和不带 phi X 对照泳道)以及流动池/文库制备效果,对 DE 结果的影响。我们研究了读取计数标准化方法对 DE 结果的影响,并表明通过总泳道计数(例如 RPKM)缩放的标准方法可能会使 DE 的估计产生偏差。我们提出了更通用的基于分位数的标准化程序,并证明了 DE 检测的改进。
我们的结果对 mRNA-Seq 实验的设计和分析具有重要的实际和方法学意义。它们强调了适当的统计方法对于标准化和 DE 推断的重要性,以考虑可能影响结果准确性的测序平台的特征。它们还揭示了在开发用于 mRNA-Seq 的统计和计算方法方面需要进一步研究。