Department of Biomedical Engineering, University of Virginia, Charlottesville, Virginia, United States of America.
PLoS Comput Biol. 2011 Mar;7(3):e1001116. doi: 10.1371/journal.pcbi.1001116. Epub 2011 Mar 31.
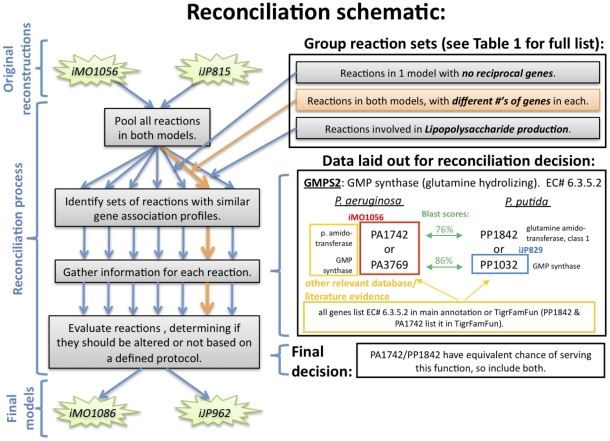
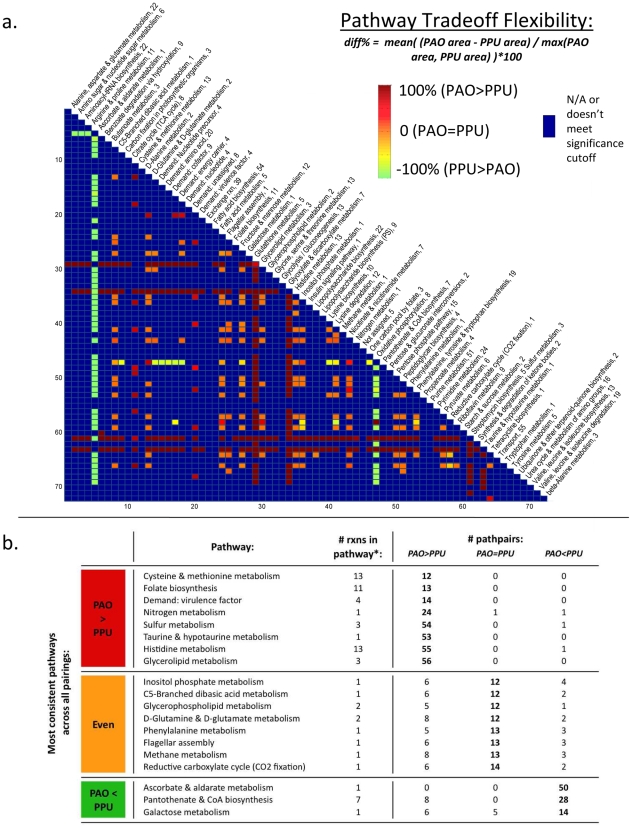
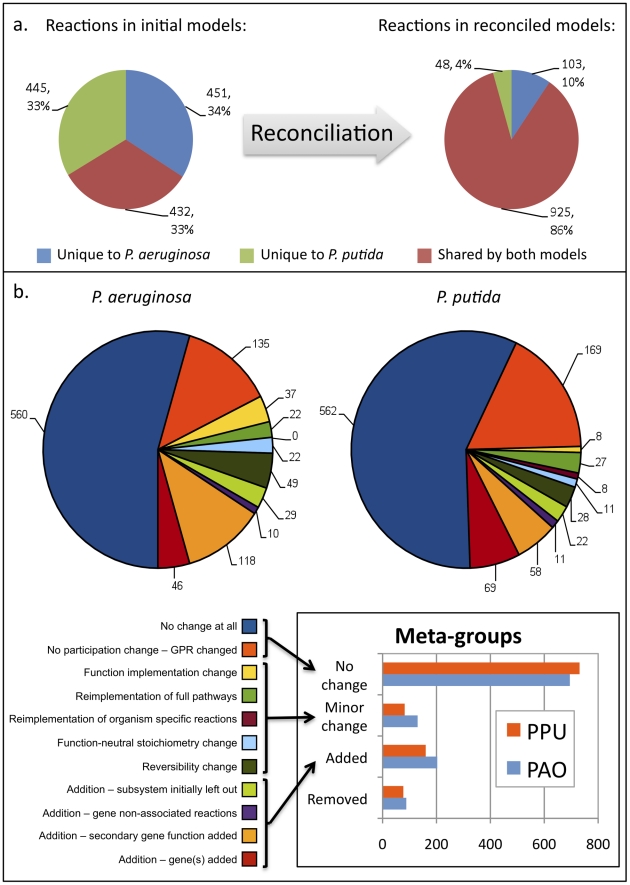
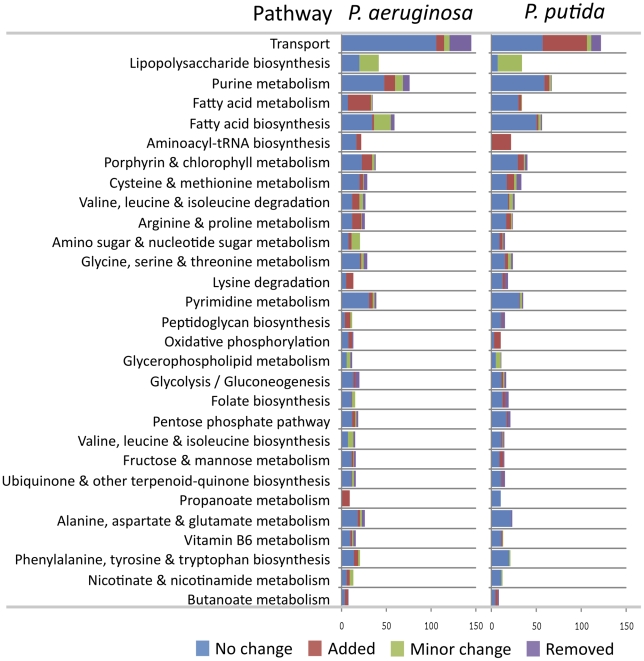
In the past decade, over 50 genome-scale metabolic reconstructions have been built for a variety of single- and multi- cellular organisms. These reconstructions have enabled a host of computational methods to be leveraged for systems-analysis of metabolism, leading to greater understanding of observed phenotypes. These methods have been sparsely applied to comparisons between multiple organisms, however, due mainly to the existence of differences between reconstructions that are inherited from the respective reconstruction processes of the organisms to be compared. To circumvent this obstacle, we developed a novel process, termed metabolic network reconciliation, whereby non-biological differences are removed from genome-scale reconstructions while keeping the reconstructions as true as possible to the underlying biological data on which they are based. This process was applied to two organisms of great importance to disease and biotechnological applications, Pseudomonas aeruginosa and Pseudomonas putida, respectively. The result is a pair of revised genome-scale reconstructions for these organisms that can be analyzed at a systems level with confidence that differences are indicative of true biological differences (to the degree that is currently known), rather than artifacts of the reconstruction process. The reconstructions were re-validated with various experimental data after reconciliation. With the reconciled and validated reconstructions, we performed a genome-wide comparison of metabolic flexibility between P. aeruginosa and P. putida that generated significant new insight into the underlying biology of these important organisms. Through this work, we provide a novel methodology for reconciling models, present new genome-scale reconstructions of P. aeruginosa and P. putida that can be directly compared at a network level, and perform a network-wide comparison of the two species. These reconstructions provide fresh insights into the metabolic similarities and differences between these important Pseudomonads, and pave the way towards full comparative analysis of genome-scale metabolic reconstructions of multiple species.
在过去的十年中,已经为多种单细胞和多细胞生物构建了超过 50 个基于基因组规模的代谢重建。这些重建使许多计算方法得以应用于代谢系统分析,从而加深了对观察到的表型的理解。然而,由于需要比较的重建之间存在差异,这些方法很少应用于多个生物体之间的比较,主要是因为这些差异是从要比较的生物体各自的重建过程中继承而来的。为了克服这一障碍,我们开发了一种新的过程,称为代谢网络协调,通过该过程可以从基于基因组规模的重建中去除非生物学差异,同时使重建尽可能真实地反映其基础生物学数据。该过程应用于对疾病和生物技术应用具有重要意义的两种生物体,即铜绿假单胞菌和恶臭假单胞菌。结果是为这两种生物体分别得到一对经过修订的基于基因组规模的重建,这些重建可以在系统水平上进行分析,并且可以确信差异是真实的生物学差异(在目前已知的程度上),而不是重建过程的人为产物。在协调后,对重建进行了各种实验数据的重新验证。在协调和验证后的重建基础上,我们对铜绿假单胞菌和恶臭假单胞菌之间的代谢灵活性进行了全基因组比较,这为这些重要生物体的基础生物学提供了重要的新见解。通过这项工作,我们提供了一种用于协调模型的新方法,提出了可以在网络水平上直接比较的铜绿假单胞菌和恶臭假单胞菌的新基于基因组规模的重建,并对这两个物种进行了网络范围的比较。这些重建为这些重要假单胞菌之间的代谢相似性和差异提供了新的见解,并为多种物种的基于基因组规模的代谢重建的全面比较分析铺平了道路。