Quantum Theory Project, University of Florida, Gainesville, Florida, United States of America.
PLoS One. 2011 Apr 25;6(4):e18868. doi: 10.1371/journal.pone.0018868.
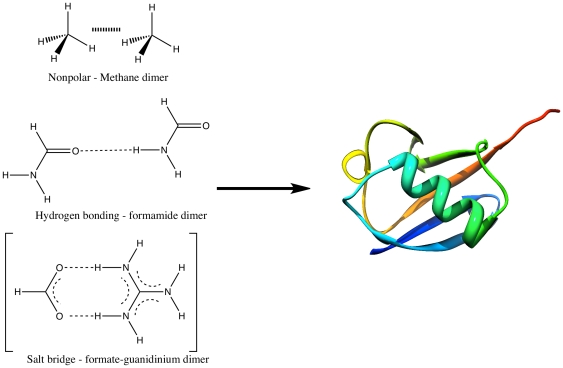
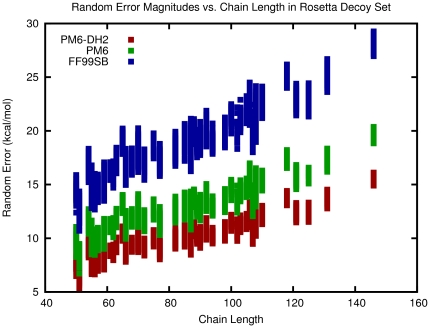
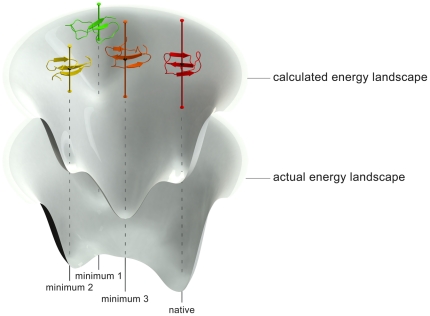
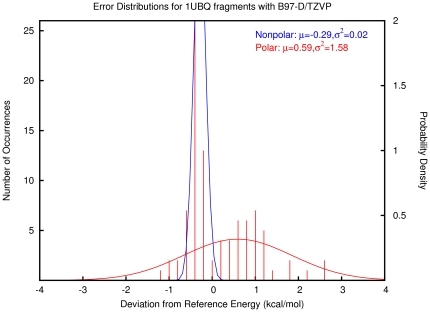
The routine prediction of three-dimensional protein structure from sequence remains a challenge in computational biochemistry. It has been intuited that calculated energies from physics-based scoring functions are able to distinguish native from nonnative folds based on previous performance with small proteins and that conformational sampling is the fundamental bottleneck to successful folding. We demonstrate that as protein size increases, errors in the computed energies become a significant problem. We show, by using error probability density functions, that physics-based scores contain significant systematic and random errors relative to accurate reference energies. These errors propagate throughout an entire protein and distort its energy landscape to such an extent that modern scoring functions should have little chance of success in finding the free energy minima of large proteins. Nonetheless, by understanding errors in physics-based score functions, they can be reduced in a post-hoc manner, improving accuracy in energy computation and fold discrimination.
从序列预测三维蛋白质结构仍然是计算生物化学中的一个挑战。人们直觉地认为,基于物理的评分函数计算出的能量能够根据小蛋白质的先前性能区分天然构象和非天然构象,并且构象采样是成功折叠的基本瓶颈。我们证明,随着蛋白质大小的增加,计算出的能量中的误差会成为一个重大问题。我们通过使用误差概率密度函数表明,基于物理的评分包含与准确参考能量相比具有显著系统和随机误差。这些误差在整个蛋白质中传播,并使其能量景观发生如此大的扭曲,以至于现代评分函数在寻找大蛋白质的自由能最小值方面几乎没有成功的机会。尽管如此,通过了解基于物理的评分函数中的误差,可以以事后的方式对其进行减少,从而提高能量计算和折叠区分的准确性。