Max Planck Research Group for Computational Genomics and Epidemiology, Max Planck Institute for Informatics, University Campus E1 4, 66123 Saarbrücken, Germany.
BMC Bioinformatics. 2011 May 9;12:141. doi: 10.1186/1471-2105-12-141.
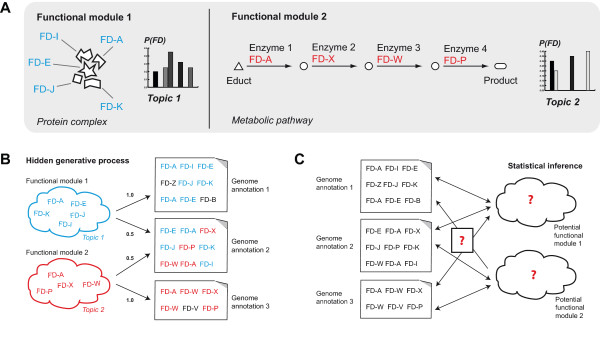
Genome and metagenome studies have identified thousands of protein families whose functions are poorly understood and for which techniques for functional characterization provide only partial information. For such proteins, the genome context can give further information about their functional context.
We describe a Bayesian method, based on a probabilistic topic model, which directly identifies functional modules of protein families. The method explores the co-occurrence patterns of protein families across a collection of sequence samples to infer a probabilistic model of arbitrarily-sized functional modules.
We show that our method identifies protein modules - some of which correspond to well-known biological processes - that are tightly interconnected with known functional interactions and are different from the interactions identified by pairwise co-occurrence. The modules are not specific to any given organism and may combine different realizations of a protein complex or pathway within different taxa.
基因组和宏基因组研究已经鉴定出数千种功能尚不清楚的蛋白质家族,而功能特征描述技术只能提供部分信息。对于此类蛋白质,其基因组背景可以提供有关其功能背景的更多信息。
我们描述了一种基于概率主题模型的贝叶斯方法,该方法可以直接识别蛋白质家族的功能模块。该方法探索了蛋白质家族在一系列序列样本中的共现模式,以推断任意大小的功能模块的概率模型。
我们表明,我们的方法可以识别蛋白质模块 - 其中一些与已知的生物过程相对应 - 这些模块与已知的功能相互作用紧密相关,并且与通过两两共现识别的相互作用不同。这些模块不限于任何特定的生物体,并且可以在不同的分类单元中组合不同的蛋白质复合物或途径的实现。