Center for Biophysics and Computational Biology, Department of Biochemistry, College of Medicine, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, United States.
J Phys Chem B. 2011 Jun 2;115(21):7029-37. doi: 10.1021/jp109631y. Epub 2011 May 11.
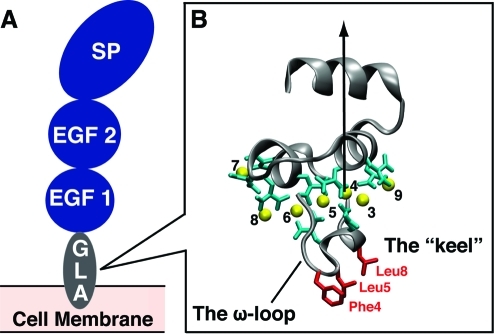
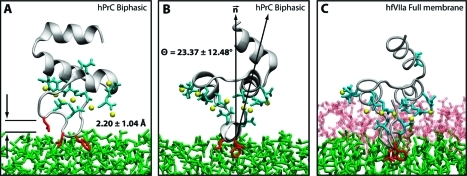
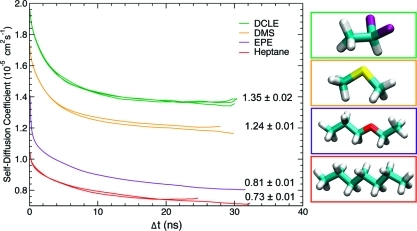
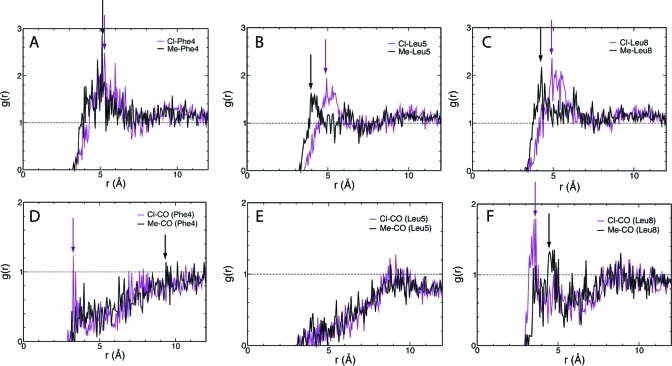
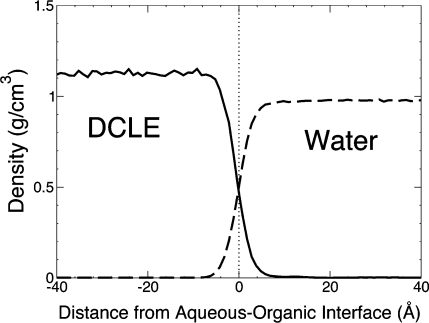
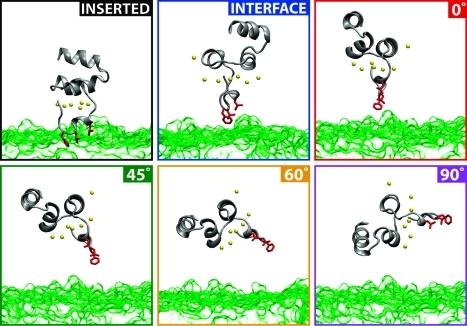
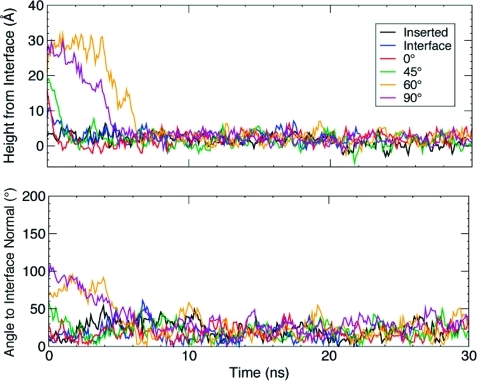
Membrane binding of peripheral proteins, mediated by specialized anchoring domains, is a crucial step for their biological function. Computational studies of membrane insertion, however, have proven challenging and largely inaccessible, due to the time scales required for the complete description of the process, mainly caused by the slow diffusion of the lipid molecules composing the membrane. Furthermore, in many cases, the nature of the membrane "anchor", i.e., the part of the protein that inserts into the membrane, is also unknown. Here, we address some of these issues by developing and employing a simplified representation of the membrane by a biphasic solvent model which we demonstrate can be used efficiently to capture and describe the process of hydrophobic insertion of membrane anchoring domains in all-atom molecular dynamics simulations. Applying the model, we have studied the insertion of the anchoring domain of a coagulation protein (the GLA domain of human protein C), starting from multiple initial configurations varying with regard to the initial orientation and height of the protein with respect to the membrane. In addition to efficiently and consistently identifying the "keel" region as the hydrophobic membrane anchor, within a few nanoseconds each configuration simulated showed a convergent height (2.20 ± 1.04 Å) and angle with respect to the interface normal (23.37 ± 12.48°). We demonstrate that the model can produce the same results as those obtained from a full representation of a membrane, in terms of both the depth of penetration and the orientation of the protein in the final membrane-bound form with an order of magnitude decrease in the required computational time compared to previous models, allowing for a more exhaustive search for the correct membrane-bound configuration.
外周蛋白通过专门的锚定域与膜结合,这是其发挥生物学功能的关键步骤。然而,由于描述整个过程所需的时间尺度,即组成膜的脂质分子的扩散非常缓慢,因此膜插入的计算研究一直具有挑战性且难以实现,在许多情况下,膜“锚”的性质,即插入膜中的蛋白质部分,也是未知的。在这里,我们通过开发和使用由双相溶剂模型表示的简化膜来解决其中的一些问题,我们证明该模型可以有效地用于捕获和描述在全原子分子动力学模拟中疏水插入膜锚定域的过程。应用该模型,我们研究了凝血蛋白(人蛋白 C 的 GLA 结构域)的锚定域从多种初始构象开始插入,这些初始构象在初始蛋白与膜的取向和高度方面有所不同。除了有效地一致地确定“龙骨”区域作为疏水区的膜锚之外,每个模拟的构象在几纳秒内都显示出收敛的高度(2.20 ± 1.04 Å)和相对于界面法线的角度(23.37 ± 12.48°)。我们证明,该模型可以与全膜表示产生相同的结果,无论是在蛋白质穿透的深度还是在最终的膜结合形式中的取向方面,与之前的模型相比,计算时间减少了一个数量级,从而可以更彻底地搜索正确的膜结合构象。