Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore, Maryland 21201, United States.
J Phys Chem B. 2011 Jun 9;115(22):7487-96. doi: 10.1021/jp202542g. Epub 2011 May 12.
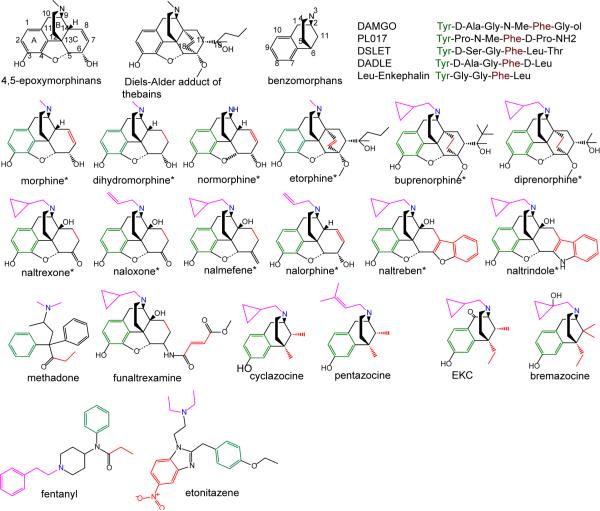
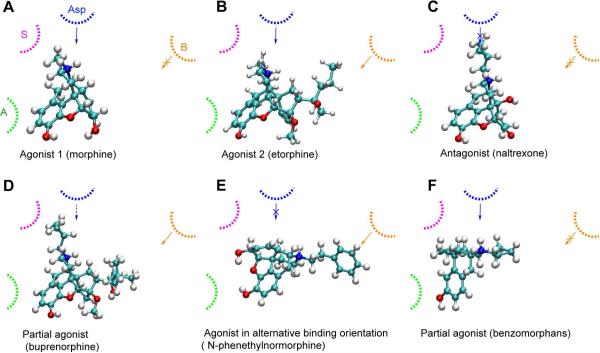
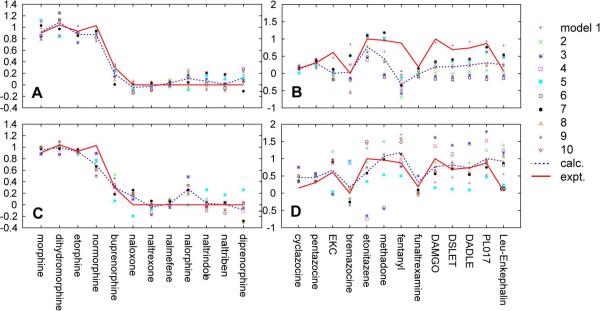
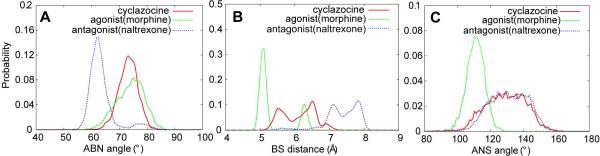
Despite being studied for over 30 years, a consensus structure-activity relationship (SAR) that encompasses the full range peptidic and nonpeptidic μ-opioid receptor ligands is still not available. To achieve a consensus SAR the Conformationally Sampled Pharmacophore (CSP) method was applied to develop a predictive model of the efficacy of μ-opioid receptor ligands. Emphasis was placed on predicting the efficacy of a wide range of agonists, partial agonists, and antagonists as well as understanding their mode of interaction with the receptor. Inclusion of all accessible conformations of each ligand, a central feature of the CSP method, enabled structural features between diverse μ-opioid receptor ligands that dictate efficacy to be identified. The models were validated against a diverse collection of peptidic and nonpeptidic ligands, including benzomorphans, fentanyl (4-anilinopiperidine), methadone (3,3-diphenylpropylamines), etonitazene (benzimidazole derivatives), funaltrexamine (C6-substituted 4,5-epoxymorphinan), and herkinorin. The model predicts (1) that interactions of ligands with the B site, as with the 19-alkyl substituents of oripavines, modulate the extent of agonism; (2) that agonists with long N-substituents, as with fentanyl and N-phenethylnormorphine, can bind in an orientation such that the N substitutent interacts with the B site that also allows the basic N-receptor Asp interaction essential for agonism; and (3) that the μ agonist herkinorin, that lacks a basic nitrogen, binds to the receptor in a manner similar to the traditional opioids via interactions mediated by water or a ion. Importantly, the proposed CSP model can be reconciled with previously published SAR models for the μ receptor.
尽管已经研究了 30 多年,但仍然没有一个包含完整的肽类和非肽类μ-阿片受体配体的共识结构-活性关系(SAR)。为了达成共识 SAR,应用构象采样药效团(CSP)方法来开发μ-阿片受体配体效能的预测模型。重点是预测广泛的激动剂、部分激动剂和拮抗剂的效能,并了解它们与受体的相互作用模式。纳入每个配体的所有可及构象是 CSP 方法的一个核心特征,这使得能够确定决定效能的不同μ-阿片受体配体之间的结构特征。该模型通过使用包括苯并吗啡烷类、芬太尼(4-苯胺基哌啶)、美沙酮(3,3-二苯基丙胺)、依托尼秦(苯并咪唑衍生物)、非那佐辛(C6-取代的 4,5-环氧吗啡烷)和海洛因醇的肽类和非肽类配体的多样数据集进行了验证。该模型预测:(1)与 19-烷基取代的阿片样物质一样,配体与 B 位的相互作用调节激动作用的程度;(2)与芬太尼和 N-苯乙基-N-去甲吗啡一样,具有长 N-取代基的激动剂可以以这样的方式结合,即 N 取代基与 B 位相互作用,这也允许碱性 N-受体天冬氨酸相互作用对激动作用至关重要;(3)μ 激动剂海洛因醇缺乏碱性氮,以类似于传统阿片类药物的方式与受体结合,通过由水或离子介导的相互作用。重要的是,所提出的 CSP 模型可以与以前发表的μ 受体 SAR 模型相协调。