Institute of Biophysical Chemistry, Center for Biomolecular Magnetic Resonance, and Frankfurt Institute for Advanced Studies, Goethe University Frankfurt am Main, Max-von-Laue-Str, 9, 60438 Frankfurt am Main, Germany.
BMC Bioinformatics. 2011 May 18;12:170. doi: 10.1186/1471-2105-12-170.
The automation of objectively selecting amino acid residue ranges for structure superpositions is important for meaningful and consistent protein structure analyses. So far there is no widely-used standard for choosing these residue ranges for experimentally determined protein structures, where the manual selection of residue ranges or the use of suboptimal criteria remain commonplace.
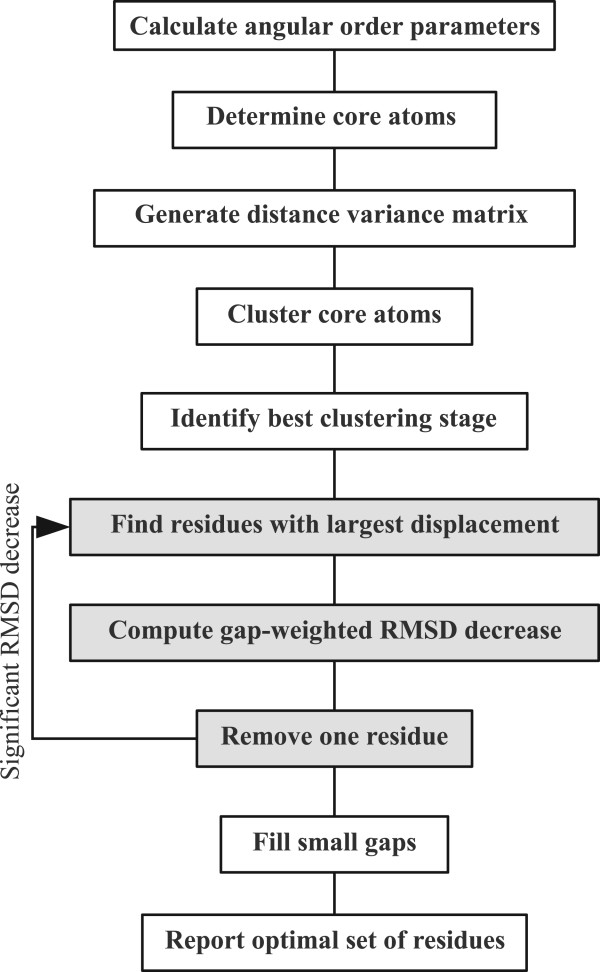
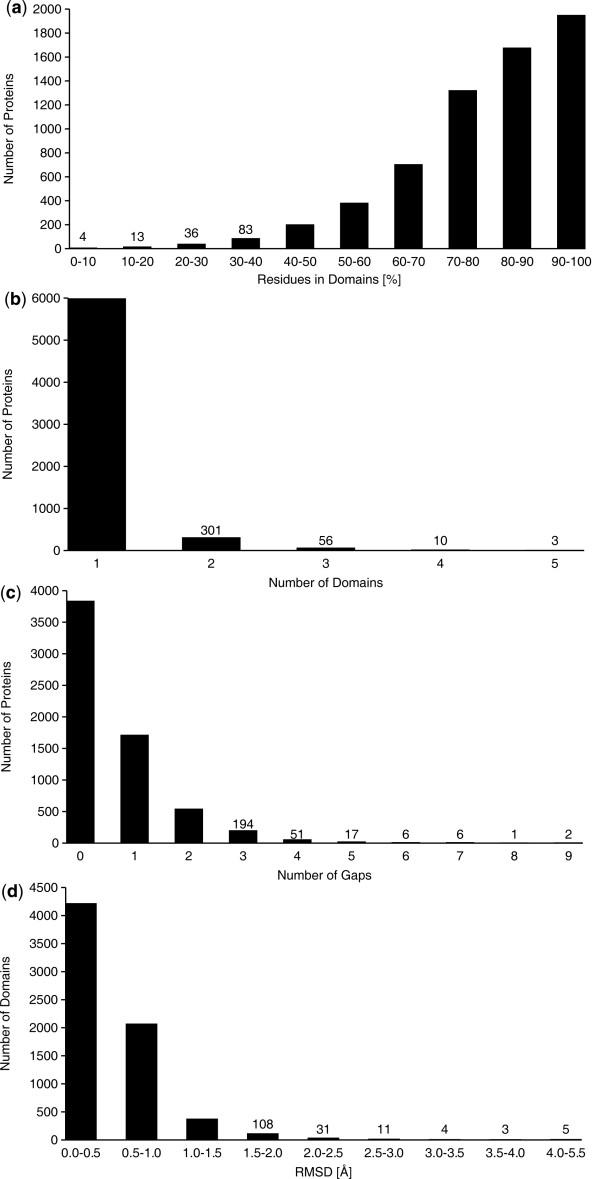
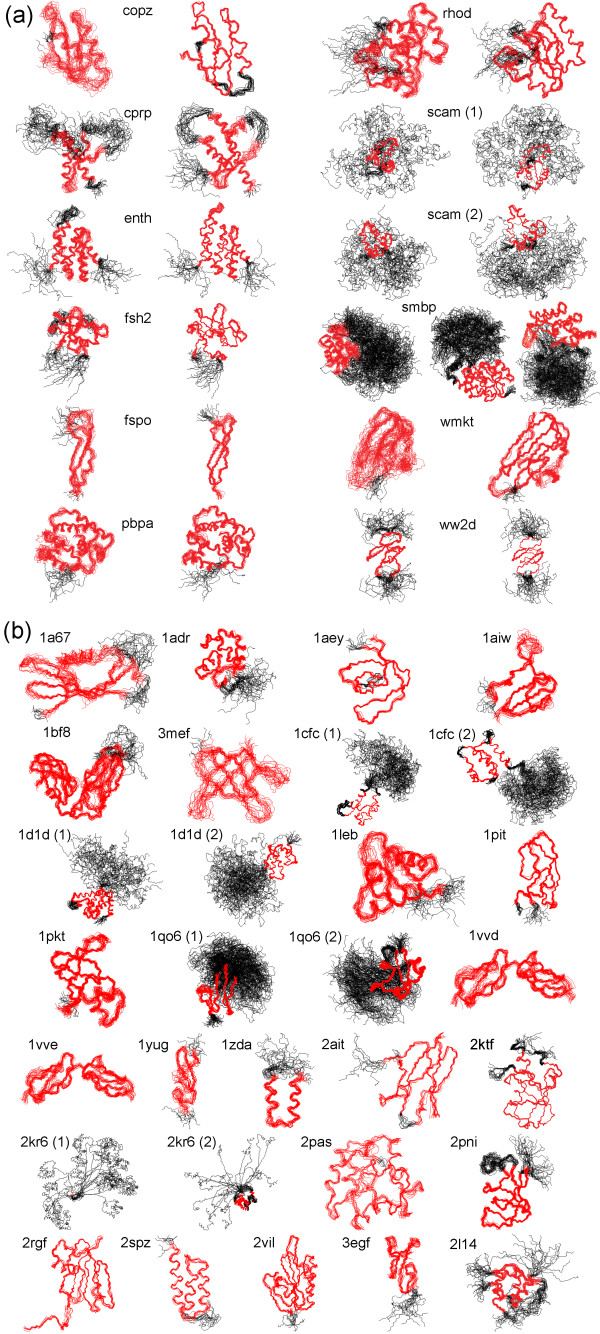
We present an automated and objective method for finding amino acid residue ranges for the superposition and analysis of protein structures, in particular for structure bundles resulting from NMR structure calculations. The method is implemented in an algorithm, CYRANGE, that yields, without protein-specific parameter adjustment, appropriate residue ranges in most commonly occurring situations, including low-precision structure bundles, multi-domain proteins, symmetric multimers, and protein complexes. Residue ranges are chosen to comprise as many residues of a protein domain that increasing their number would lead to a steep rise in the RMSD value. Residue ranges are determined by first clustering residues into domains based on the distance variance matrix, and then refining for each domain the initial choice of residues by excluding residues one by one until the relative decrease of the RMSD value becomes insignificant. A penalty for the opening of gaps favours contiguous residue ranges in order to obtain a result that is as simple as possible, but not simpler. Results are given for a set of 37 proteins and compared with those of commonly used protein structure validation packages. We also provide residue ranges for 6351 NMR structures in the Protein Data Bank.
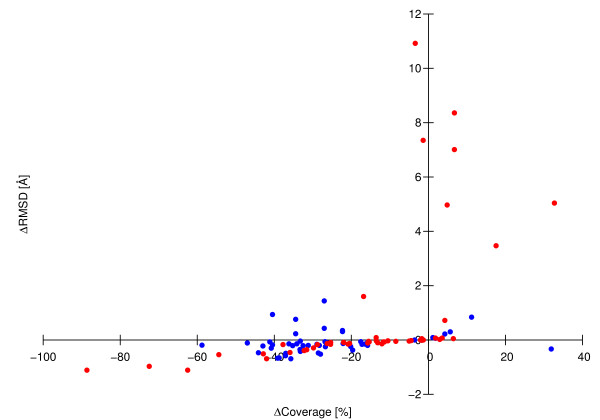
The CYRANGE method is capable of automatically determining residue ranges for the superposition of protein structure bundles for a large variety of protein structures. The method correctly identifies ordered regions. Global structure superpositions based on the CYRANGE residue ranges allow a clear presentation of the structure, and unnecessary small gaps within the selected ranges are absent. In the majority of cases, the residue ranges from CYRANGE contain fewer gaps and cover considerably larger parts of the sequence than those from other methods without significantly increasing the RMSD values. CYRANGE thus provides an objective and automatic method for standardizing the choice of residue ranges for the superposition of protein structures.
客观选择氨基酸残基范围进行结构叠加对于有意义且一致的蛋白质结构分析非常重要。到目前为止,对于实验确定的蛋白质结构,还没有广泛使用的标准来选择这些残基范围,手动选择残基范围或使用非最佳标准仍然很常见。
我们提出了一种自动且客观的方法,用于为蛋白质结构的叠加和分析寻找氨基酸残基范围,特别是对于 NMR 结构计算产生的结构束。该方法在算法 CYRANGE 中实现,在大多数常见情况下,包括低精度结构束、多域蛋白、对称多聚体和蛋白质复合物,无需蛋白质特定参数调整,就可以生成合适的残基范围。残基范围的选择是为了包含尽可能多的蛋白质结构域残基,如果增加其数量会导致 RMSD 值急剧上升。残基范围是通过首先根据距离方差矩阵将残基聚类为结构域,然后针对每个结构域逐个排除残基来细化初始残基选择,直到 RMSD 值的相对降低变得不显著为止。为了获得尽可能简单但不是更简单的结果,对打开间隙的惩罚有利于连续的残基范围。结果针对一组 37 个蛋白质进行了给出,并与常用的蛋白质结构验证包的结果进行了比较。我们还为蛋白质数据库中的 6351 个 NMR 结构提供了残基范围。
CYRANGE 方法能够自动确定用于各种蛋白质结构叠加的蛋白质结构束的残基范围。该方法正确识别有序区域。基于 CYRANGE 残基范围的全局结构叠加允许清晰地呈现结构,并且所选范围内不存在不必要的小间隙。在大多数情况下,与其他方法相比,CYRANGE 的残基范围包含更少的间隙,并且覆盖了序列的更大部分,而不会显著增加 RMSD 值。因此,CYRANGE 为蛋白质结构叠加中残基范围的选择提供了一种客观且自动的方法。