Center for Health Informatics and Bioinformatics, New York University School of Medicine, New York, NY 10016, USA.
Biol Direct. 2011 May 18;6:25. doi: 10.1186/1745-6150-6-25.
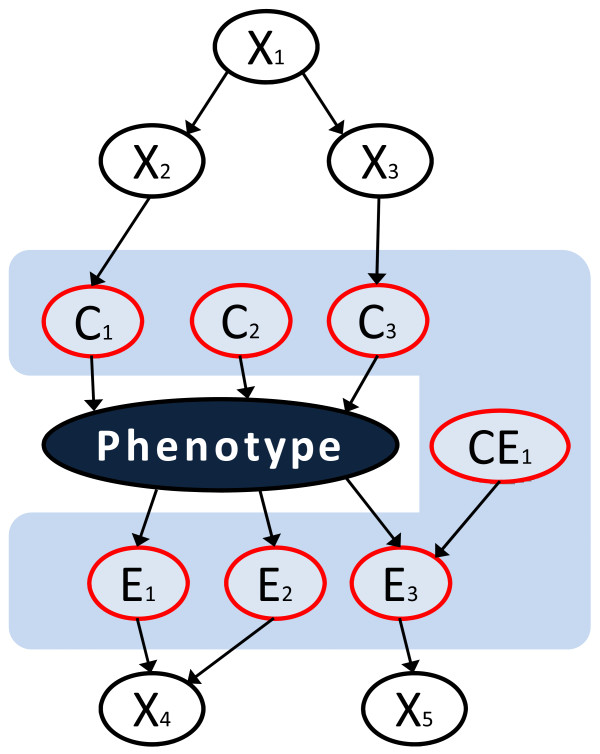
GWAS owe their popularity to the expectation that they will make a major impact on diagnosis, prognosis and management of disease by uncovering genetics underlying clinical phenotypes. The dominant paradigm in GWAS data analysis so far consists of extensive reliance on methods that emphasize contribution of individual SNPs to statistical association with phenotypes. Multivariate methods, however, can extract more information by considering associations of multiple SNPs simultaneously. Recent advances in other genomics domains pinpoint multivariate causal graph-based inference as a promising principled analysis framework for high-throughput data. Designed to discover biomarkers in the local causal pathway of the phenotype, these methods lead to accurate and highly parsimonious multivariate predictive models. In this paper, we investigate the applicability of causal graph-based method TIE* to analysis of GWAS data. To test the utility of TIE*, we focus on anti-CCP positive rheumatoid arthritis (RA) GWAS datasets, where there is a general consensus in the community about the major genetic determinants of the disease.
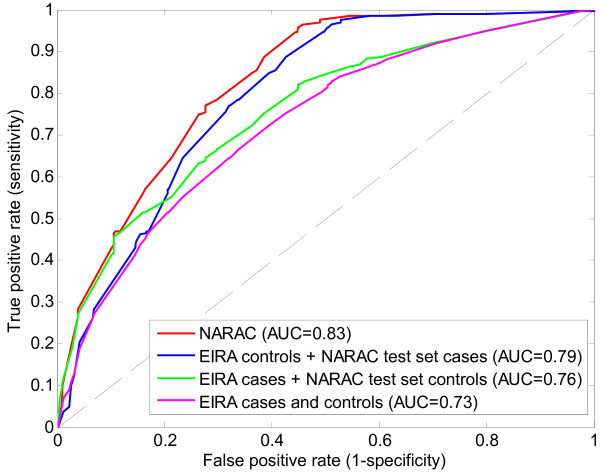
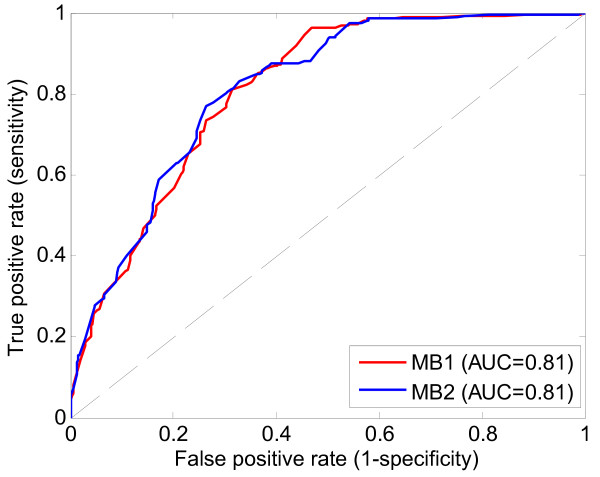
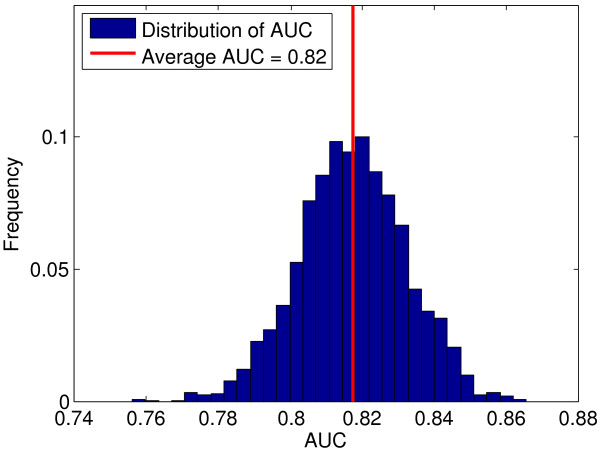
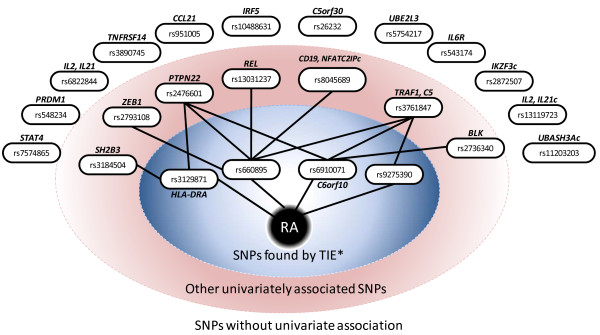
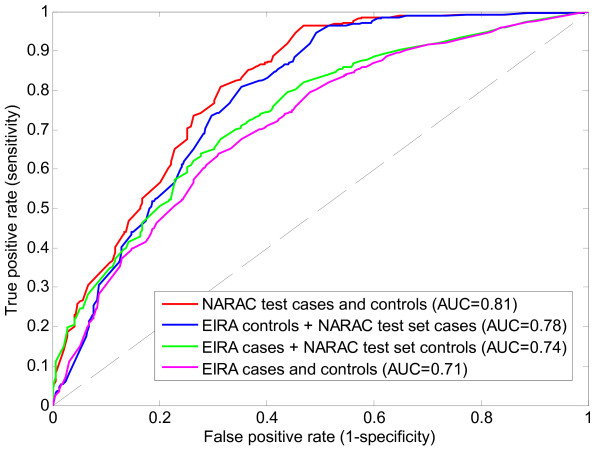
Application of TIE* to the North American Rheumatoid Arthritis Cohort (NARAC) GWAS data results in six SNPs, mostly from the MHC locus. Using these SNPs we develop two predictive models that can classify cases and disease-free controls with an accuracy of 0.81 area under the ROC curve, as verified in independent testing data from the same cohort. The predictive performance of these models generalizes reasonably well to Swedish subjects from the closely related but not identical Epidemiological Investigation of Rheumatoid Arthritis (EIRA) cohort with 0.71-0.78 area under the ROC curve. Moreover, the SNPs identified by the TIE* method render many other previously known SNP associations conditionally independent of the phenotype.
Our experiments demonstrate that application of TIE* captures maximum amount of genetic information about RA in the data and recapitulates the major consensus findings about the genetic factors of this disease. In addition, TIE* yields reproducible markers and signatures of RA. This suggests that principled multivariate causal and predictive framework for GWAS analysis empowers the community with a new tool for high-quality and more efficient discovery.
This article was reviewed by Prof. Anthony Almudevar, Dr. Eugene V. Koonin, and Prof. Marianthi Markatou.
全基因组关联研究 (GWAS) 之所以受到关注,是因为它们有望通过揭示临床表型背后的遗传基础,对疾病的诊断、预后和治疗产生重大影响。到目前为止,GWAS 数据分析的主导范式主要依赖于强调个体 SNP 对与表型统计关联贡献的方法。然而,多变量方法可以通过同时考虑多个 SNP 的关联来提取更多信息。其他基因组学领域的最新进展指出,基于多变量因果图的推断是一种有前途的高通量数据分析原则性分析框架。这些方法旨在发现表型局部因果途径中的生物标志物,从而导致准确且高度简约的多变量预测模型。在本文中,我们研究了基于因果图的方法 TIE在 GWAS 数据分析中的适用性。为了测试 TIE的实用性,我们专注于抗 CCP 阳性类风湿关节炎 (RA) GWAS 数据集,该领域的研究社区普遍认为该疾病的主要遗传决定因素。
将 TIE应用于北美类风湿关节炎队列 (NARAC) GWAS 数据可得到六个 SNP,主要来自 MHC 基因座。使用这些 SNP,我们开发了两个可以以 0.81ROC 曲线下面积分类病例和无病对照的预测模型,在来自同一队列的独立测试数据中得到验证。这些模型的预测性能在遗传上与密切相关但不相同的流行病学调查类风湿关节炎 (EIRA) 队列中的瑞典受试者具有很好的通用性,ROC 曲线下面积为 0.71-0.78。此外,TIE方法识别的 SNP 使许多其他先前已知的 SNP 关联条件独立于表型。
我们的实验表明,TIE应用程序捕获了数据中有关 RA 的最大遗传信息量,并再现了该疾病遗传因素的主要共识发现。此外,TIE产生了可重复的 RA 标记和特征。这表明,GWAS 分析的原则性多变量因果和预测框架为社区提供了一种新的工具,用于进行高质量、更高效的发现。
本文由 Prof. Anthony Almudevar、Dr. Eugene V. Koonin 和 Prof. Marianthi Markatou 审稿。