Genome Sciences Centre, BC Cancer Agency, Vancouver, British Columbia, Canada.
PLoS One. 2011;6(5):e19838. doi: 10.1371/journal.pone.0019838. Epub 2011 May 13.
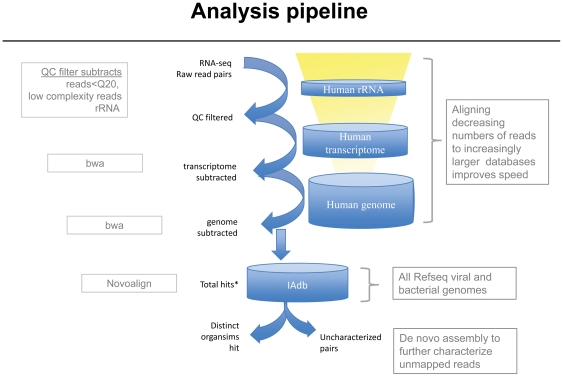
Massively parallel sequencing technology now provides the opportunity to sample the transcriptome of a given tissue comprehensively. Transcripts at only a few copies per cell are readily detectable, allowing the discovery of low abundance viral and bacterial transcripts in human tissue samples. Here we describe an approach for mining large sequence data sets for the presence of microbial sequences. Further, we demonstrate the sensitivity of this approach by sequencing human RNA-seq libraries spiked with decreasing amounts of an RNA-virus. At a modest depth of sequencing, viral transcripts can be detected at frequencies less than 1 in 1,000,000. With current sequencing platforms approaching outputs of one billion reads per run, this is a highly sensitive method for detecting putative infectious agents associated with human tissues.
大规模平行测序技术现在提供了全面采样特定组织转录组的机会。每个细胞中只有几个拷贝的转录本很容易被检测到,这使得在人类组织样本中发现低丰度的病毒和细菌转录本成为可能。在这里,我们描述了一种用于挖掘大型序列数据集以发现微生物序列的方法。此外,我们通过对用逐渐减少量的 RNA 病毒进行 Spike 的人类 RNA-seq 文库进行测序,证明了这种方法的灵敏度。在适度的测序深度下,病毒转录本的检测频率可以低至每 100 万分之一。随着当前测序平台的输出接近每个运行 10 亿个读数,这是一种非常敏感的方法,可用于检测与人体组织相关的潜在传染性病原体。