Computer Science and Engineering Department, Michigan State University, East Lansing, USA.
BMC Bioinformatics. 2011 May 24;12:198. doi: 10.1186/1471-2105-12-198.
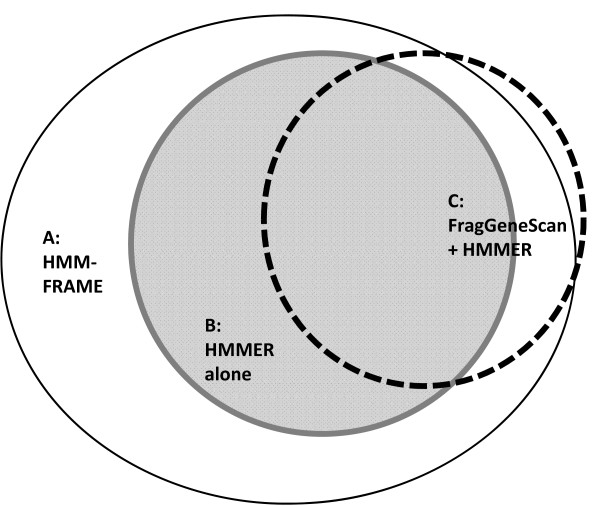
Protein domain classification is an important step in metagenomic annotation. The state-of-the-art method for protein domain classification is profile HMM-based alignment. However, the relatively high rates of insertions and deletions in homopolymer regions of pyrosequencing reads create frameshifts, causing conventional profile HMM alignment tools to generate alignments with marginal scores. This makes error-containing gene fragments unclassifiable with conventional tools. Thus, there is a need for an accurate domain classification tool that can detect and correct sequencing errors.
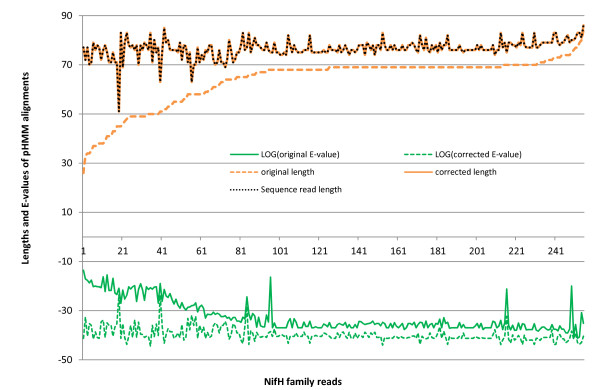
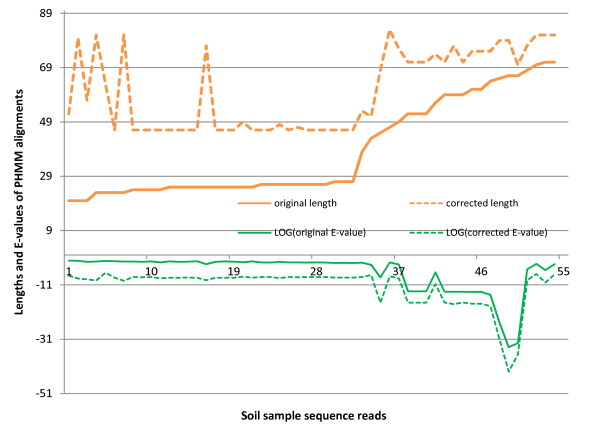
We introduce HMM-FRAME, a protein domain classification tool based on an augmented Viterbi algorithm that can incorporate error models from different sequencing platforms. HMM-FRAME corrects sequencing errors and classifies putative gene fragments into domain families. It achieved high error detection sensitivity and specificity in a data set with annotated errors. We applied HMM-FRAME in Targeted Metagenomics and a published metagenomic data set. The results showed that our tool can correct frameshifts in error-containing sequences, generate much longer alignments with significantly smaller E-values, and classify more sequences into their native families.
HMM-FRAME provides a complementary protein domain classification tool to conventional profile HMM-based methods for data sets containing frameshifts. Its current implementation is best used for small-scale metagenomic data sets. The source code of HMM-FRAME can be downloaded at http://www.cse.msu.edu/~zhangy72/hmmframe/ and at https://sourceforge.net/projects/hmm-frame/.
蛋白质结构域分类是宏基因组注释的重要步骤。基于轮廓隐马尔可夫模型(profile HMM)比对的方法是蛋白质结构域分类的最新技术。然而,焦磷酸测序reads 中的同源聚合物区域的插入和缺失率相对较高,导致传统的基于轮廓 HMM 的比对工具生成得分较低的比对结果。这使得含有错误的基因片段无法使用传统工具进行分类。因此,需要一种能够检测和纠正测序错误的准确结构域分类工具。
我们引入了 HMM-FRAME,这是一种基于增强维特比算法的蛋白质结构域分类工具,能够整合来自不同测序平台的错误模型。HMM-FRAME 能够纠正测序错误并将假定的基因片段分类到结构域家族中。在带有注释错误的数据集中,它具有较高的错误检测灵敏度和特异性。我们将 HMM-FRAME 应用于靶向宏基因组学和已发表的宏基因组数据集。结果表明,我们的工具能够纠正含有错误的序列中的移码,生成具有显著更小 E 值的更长比对结果,并将更多的序列分类到其天然家族中。
HMM-FRAME 为含有移码的数据集提供了一种与传统基于轮廓 HMM 的方法互补的蛋白质结构域分类工具。它的当前实现最适合于小规模的宏基因组数据集。HMM-FRAME 的源代码可以在以下网址下载:http://www.cse.msu.edu/~zhangy72/hmmframe/ 和 https://sourceforge.net/projects/hmm-frame/。