Department of Biomedical Informatics, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA.
BMC Bioinformatics. 2011 May 27;12:213. doi: 10.1186/1471-2105-12-213.
Studies integrating transcriptomic data with proteomic data can illuminate the proteome more clearly than either separately. Integromic studies can deepen understanding of the dynamic complex regulatory relationship between the transcriptome and the proteome. Integrating these data dictates a reliable mapping between the identifier nomenclature resultant from the two high-throughput platforms. However, this kind of analysis is well known to be hampered by lack of standardization of identifier nomenclature among proteins, genes, and microarray probe sets. Therefore data integration may also play a role in critiquing the fallible gene identifications that both platforms emit.
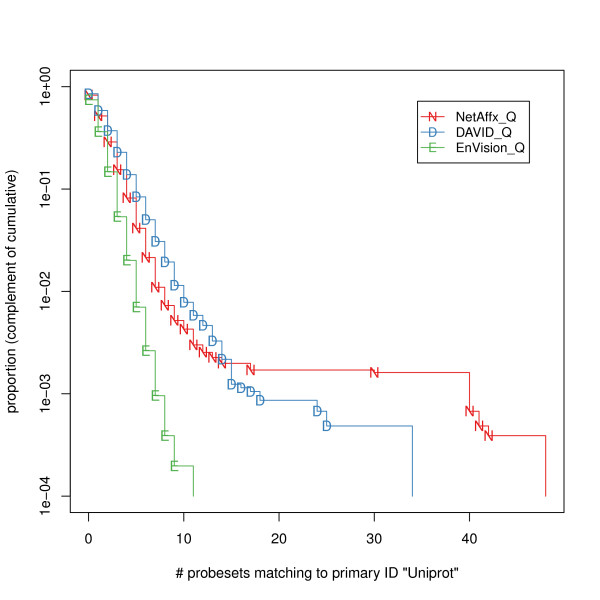
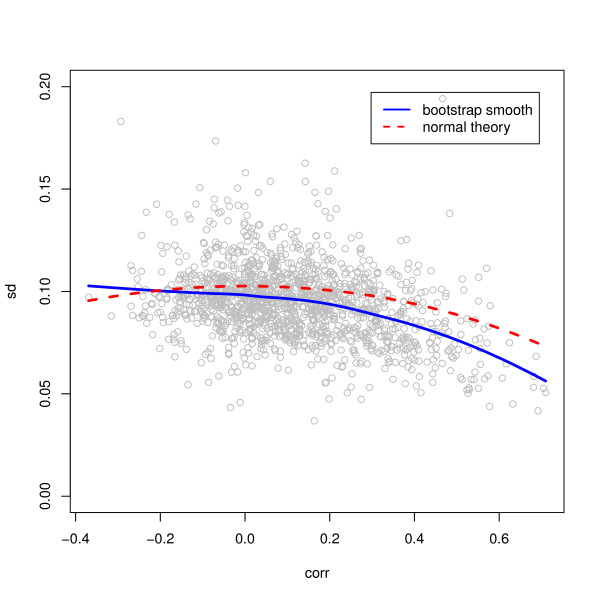
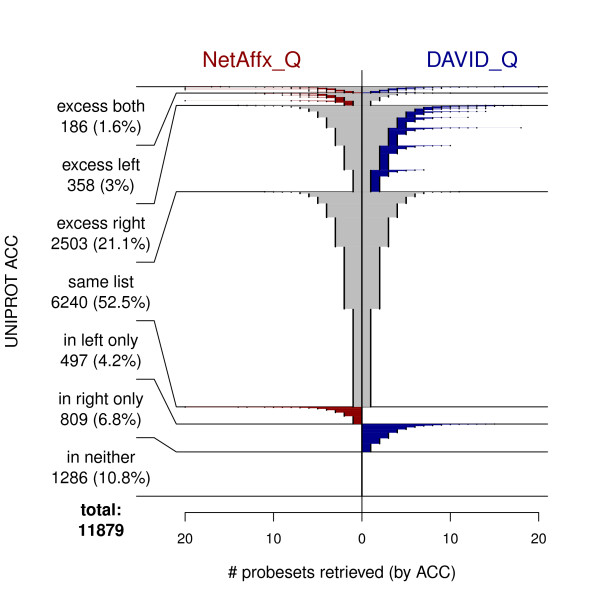
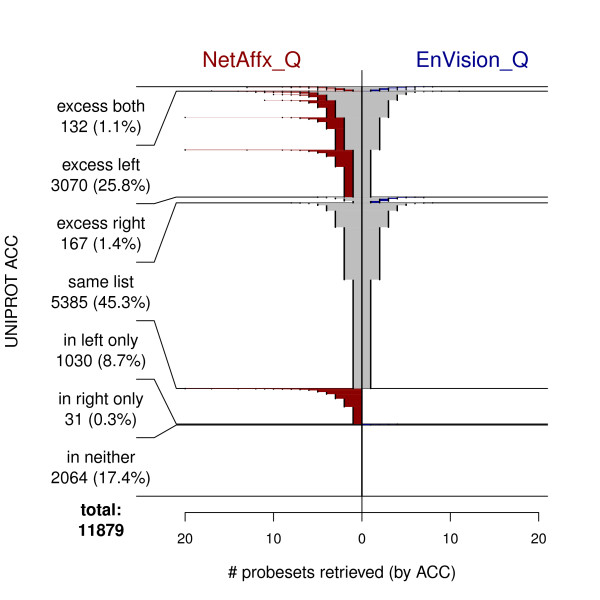
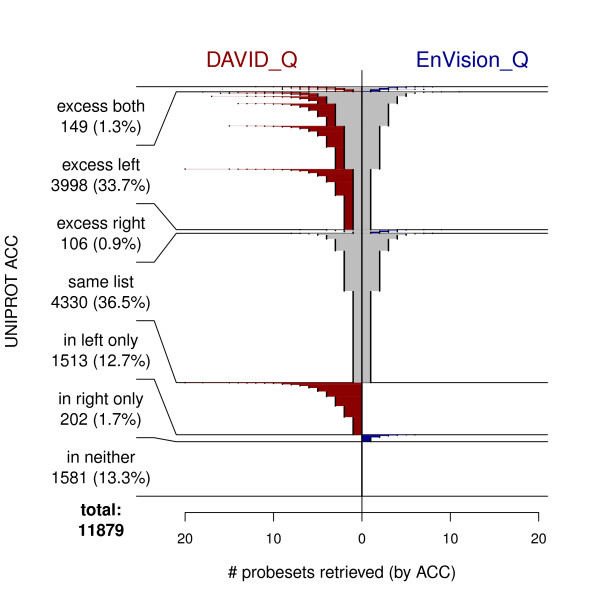
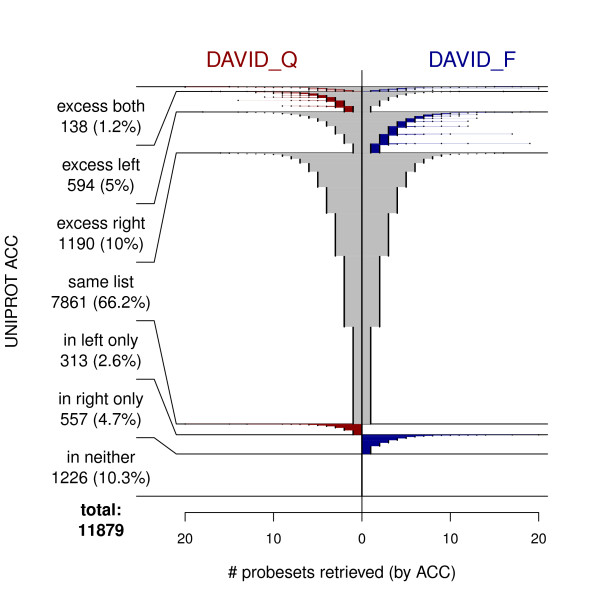
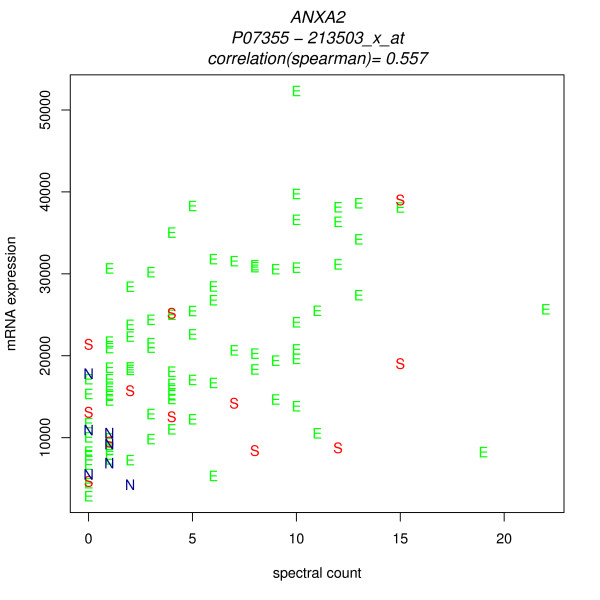
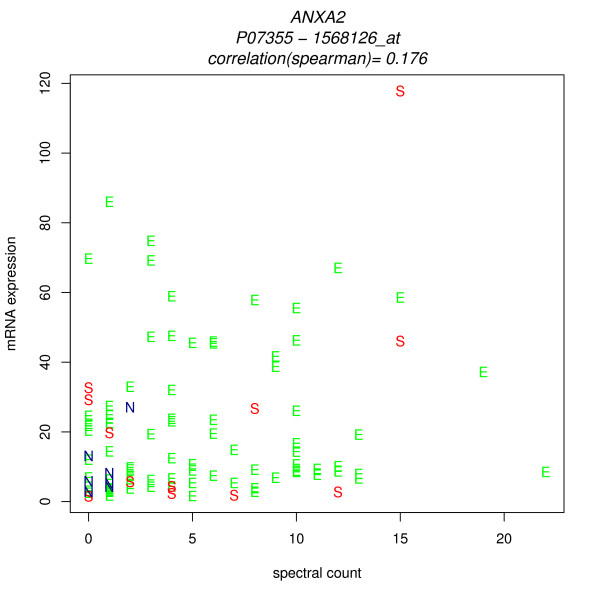
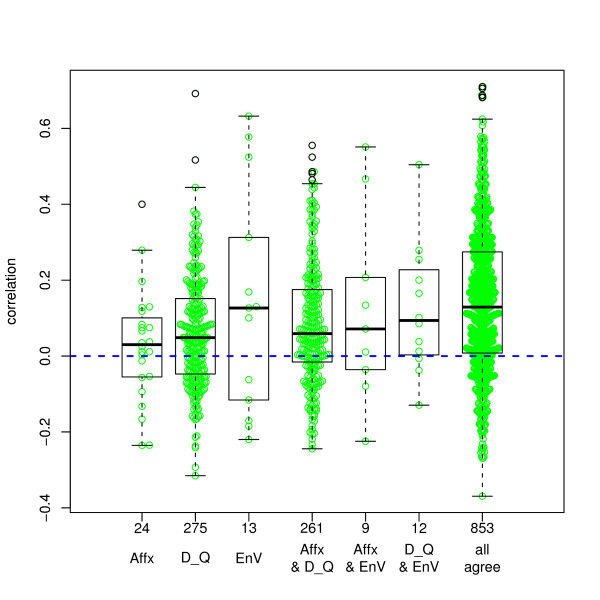
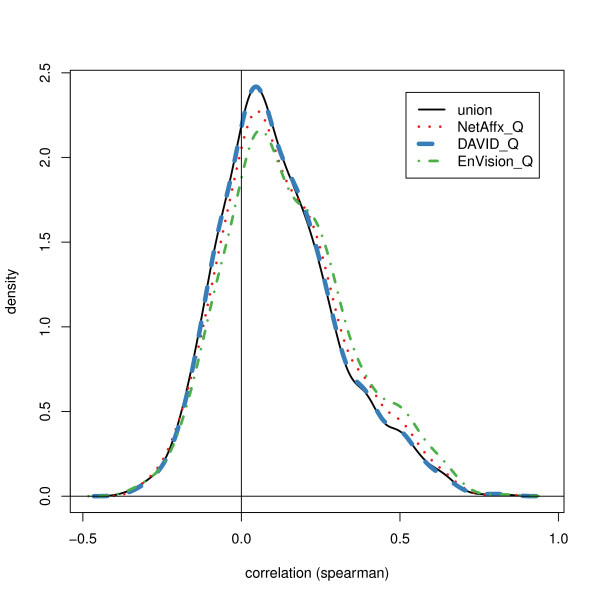
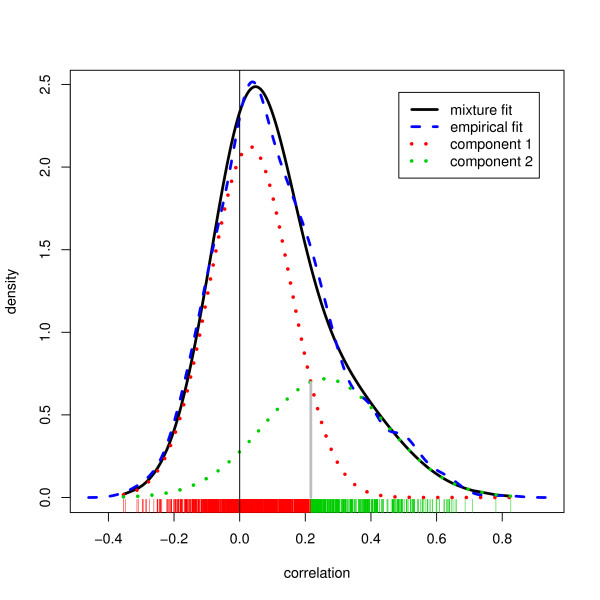
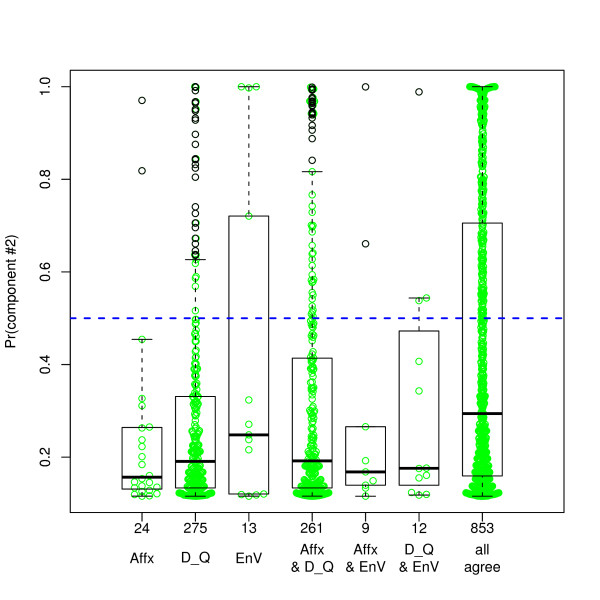
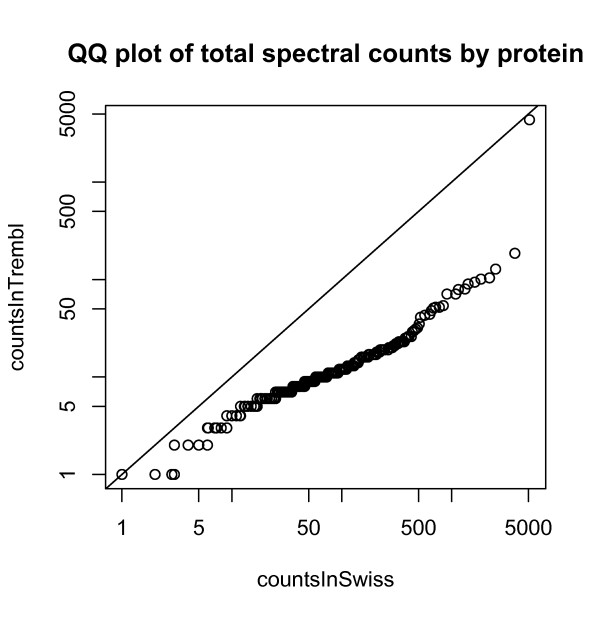
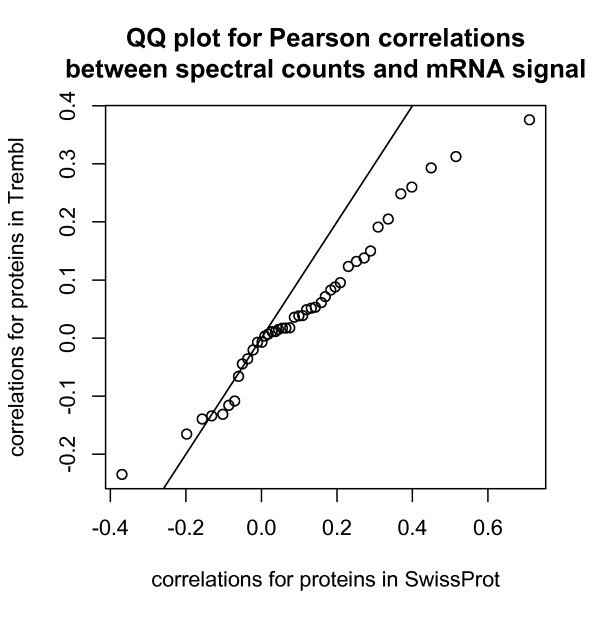
We compared three freely available internet-based identifier mapping resources for mapping UniProt accessions (ACCs) to Affymetrix probesets identifications (IDs): DAVID, EnVision, and NetAffx. Liquid chromatography-tandem mass spectrometry analyses of 91 endometrial cancer and 7 noncancer samples generated 11,879 distinct ACCs. For each ACC, we compared the retrieval sets of probeset IDs from each mapping resource. We confirmed a high level of discrepancy among the mapping resources. On the same samples, mRNA expression was available. Therefore, to evaluate the quality of each ACC-to-probeset match, we calculated proteome-transcriptome correlations, and compared the resources presuming that better mapping of identifiers should generate a higher proportion of mapped pairs with strong inter-platform correlations. A mixture model for the correlations fitted well and supported regression analysis, providing a window into the performance of the mapping resources. The resources have added and dropped matches over two years, but their overall performance has not changed.
The methods presented here serve to achieve concrete context-specific insight, to support well-informed decisions in choosing an ID mapping strategy for "omic" data merging.
将转录组数据与蛋白质组数据整合的研究比单独分析两者能更清楚地阐明蛋白质组。整合组学研究可以加深对转录组和蛋白质组之间动态复杂调控关系的理解。整合这些数据需要在两种高通量平台产生的标识符命名法之间建立可靠的映射。然而,这种分析由于蛋白质、基因和微阵列探针集标识符命名法缺乏标准化而受到阻碍。因此,数据整合也可能在批评两个平台产生的不可靠基因识别方面发挥作用。
我们比较了三种免费的基于互联网的标识符映射资源,用于将 UniProt 访问号(ACCs)映射到 Affymetrix 探针集标识符(IDs):DAVID、EnVision 和 NetAffx。对 91 个子宫内膜癌和 7 个非癌样本进行液相色谱-串联质谱分析,生成了 11879 个独特的 ACCs。对于每个 ACC,我们比较了每个映射资源的探针集 ID 的检索集。我们确认了映射资源之间存在高度差异。在相同的样本上,我们可以获得 mRNA 表达信息。因此,为了评估每个 ACC 与探针集匹配的质量,我们计算了蛋白质组与转录组的相关性,并根据资源假设标识符的更好映射应该生成具有较强平台间相关性的映射对的更高比例来比较资源。相关性的混合模型拟合良好,并支持回归分析,为映射资源的性能提供了一个窗口。这些资源在两年内增加和删除了匹配项,但整体性能没有改变。
这里提出的方法提供了具体的上下文特定的见解,支持在选择“组学”数据合并的标识符映射策略时做出明智的决策。