Department of Microbiology and Immunology, Albert Einstein College of Medicine, Bronx, New York, United States of America.
PLoS One. 2011;6(5):e20183. doi: 10.1371/journal.pone.0020183. Epub 2011 May 27.
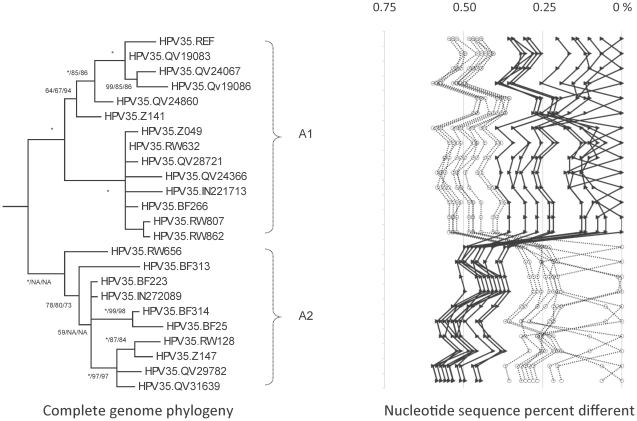
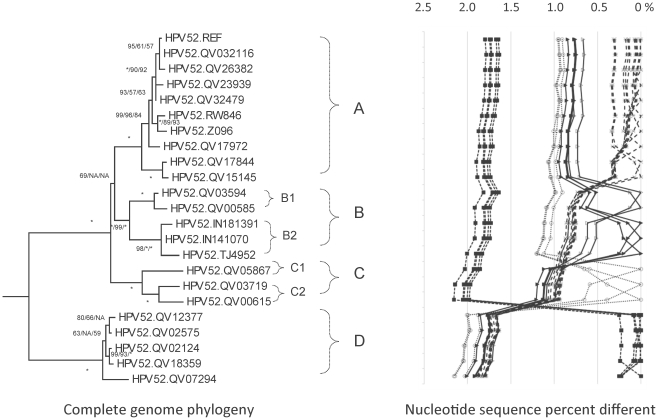
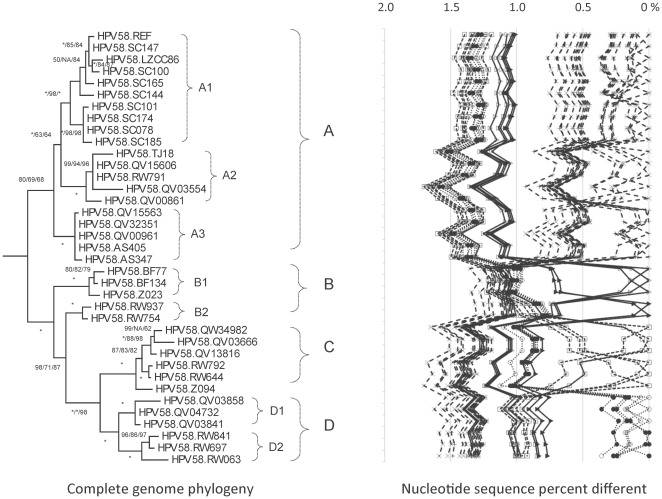
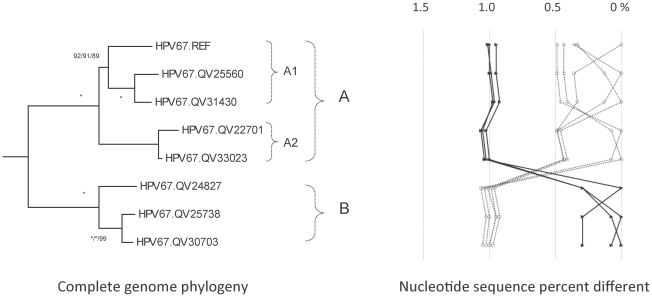
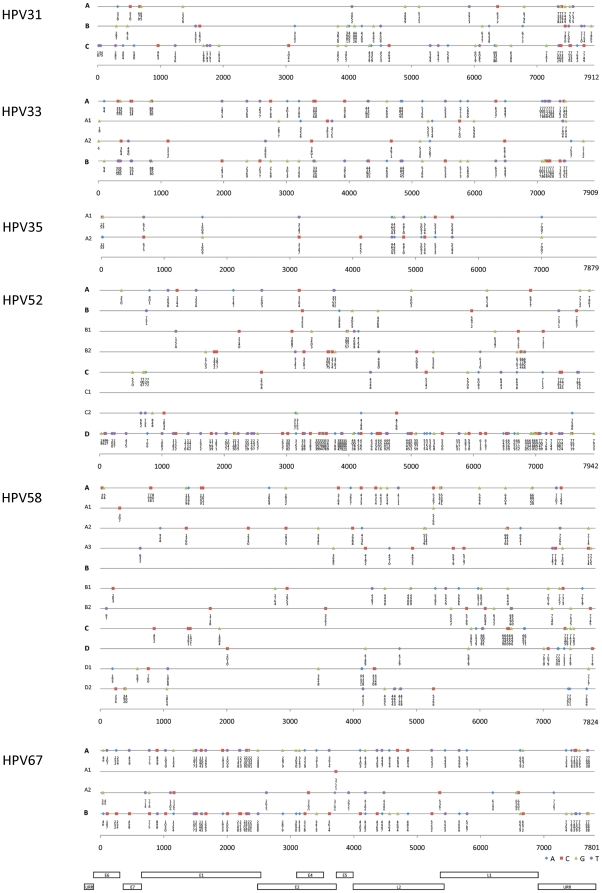
Human papillomavirus 16 (HPV16) species group (alpha-9) of the Alphapapillomavirus genus contains HPV16, HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. These HPVs account for 75% of invasive cervical cancers worldwide. Viral variants of these HPVs differ in evolutionary history and pathogenicity. Moreover, a comprehensive nomenclature system for HPV variants is lacking, limiting comparisons between studies.
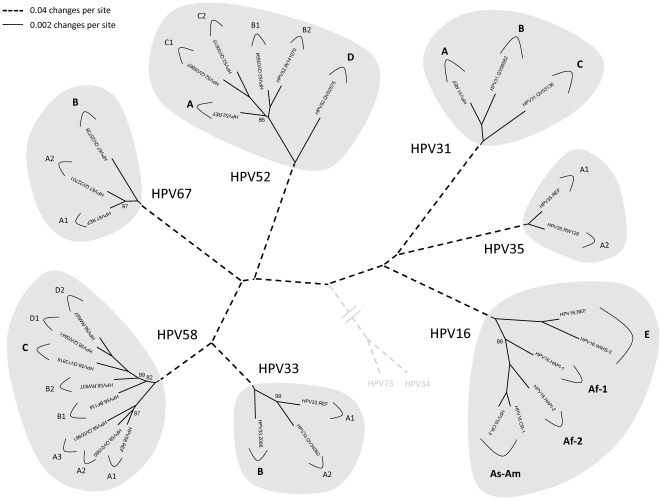
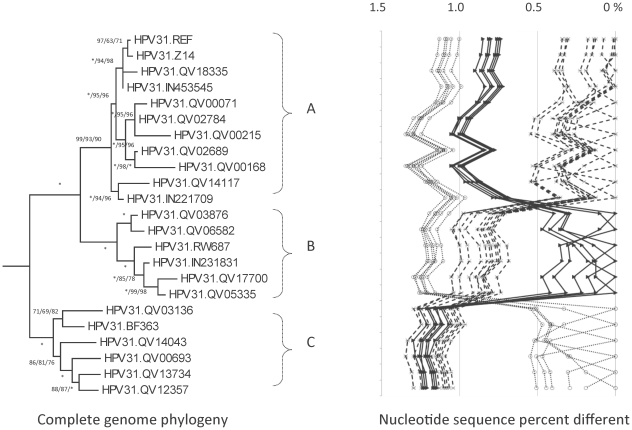
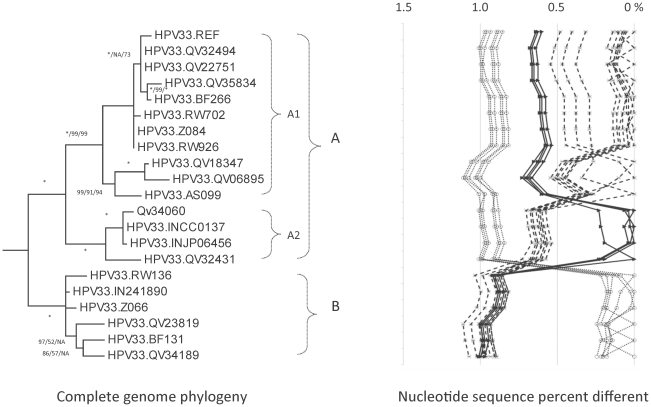
DNA from cervical samples previously characterized for HPV type were obtained from multiple geographic regions to screen for novel variants. The complete 8 kb genomes of 120 variants representing the major and minor lineages of the HPV16-related alpha-9 HPV types were sequenced to capture maximum viral heterogeneity. Viral evolution was characterized by constructing phylogenic trees based on complete genomes using multiple algorithms. Maximal and viral region specific divergence was calculated by global and pairwise alignments. Variant lineages were classified and named using an alphanumeric system; the prototype genome was assigned to the A lineage for all types.
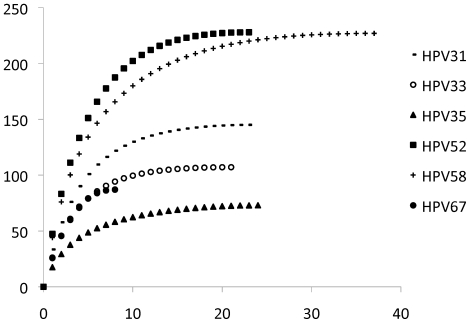
The range of genome-genome sequence heterogeneity varied from 0.6% for HPV35 to 2.2% for HPV52 and included 1.4% for HPV31, 1.1% for HPV33, 1.7% for HPV58 and 1.1% for HPV67. Nucleotide differences of approximately 1.0% - 10.0% and 0.5%-1.0% of the complete genomes were used to define variant lineages and sublineages, respectively. Each gene/region differs in sequence diversity, from most variable to least variable: noncoding region 1 (NCR1) /noncoding region 2 (NCR2) >upstream regulatory region (URR)> E6/E7 > E2/L2 > E1/L1.
These data define maximum viral genomic heterogeneity of HPV16-related alpha-9 HPV variants. The proposed nomenclature system facilitates the comparison of variants across epidemiological studies. Sequence diversity and phylogenies of this clinically important group of HPVs provides the basis for further studies of discrete viral evolution, epidemiology, pathogenesis and preventative/therapeutic interventions.
人乳头瘤病毒 16(HPV16)种组(阿尔法-9)属于阿尔法乳头瘤病毒属,包含 HPV16、HPV31、HPV33、HPV35、HPV52、HPV58 和 HPV67。这些 HPV 占全球侵袭性宫颈癌的 75%。这些 HPV 的病毒变体在进化史和致病性上有所不同。此外,HPV 变体缺乏全面的命名系统,限制了研究之间的比较。
从多个地理区域获得了先前针对 HPV 类型进行特征描述的宫颈样本中的 DNA,以筛选新的变体。对代表 HPV16 相关 alpha-9 HPV 型主要和次要谱系的 120 种变体的完整 8 kb 基因组进行测序,以捕获最大的病毒异质性。通过使用多种算法基于完整基因组构建系统发育树来描述病毒进化。通过全局和成对比对计算最大和病毒区域特异性分歧。使用字母数字系统对变体谱系进行分类和命名;为所有类型分配原型基因组到 A 谱系。
基因组-基因组序列异质性的范围从 HPV35 的 0.6%到 HPV52 的 2.2%不等,包括 HPV31 的 1.4%、HPV33 的 1.1%、HPV58 的 1.7%和 HPV67 的 1.1%。大约 1.0% - 10.0%和 0.5%-1.0%的完整基因组的核苷酸差异分别用于定义变体谱系和亚谱系。每个基因/区域的序列多样性不同,从最可变到最不可变:非编码区 1(NCR1)/非编码区 2(NCR2)>上游调控区(URR)> E6/E7 > E2/L2 > E1/L1。
这些数据定义了 HPV16 相关 alpha-9 HPV 变体的最大病毒基因组异质性。所提出的命名系统有助于在流行病学研究中比较变体。这组具有临床重要性的 HPV 的序列多样性和系统发育为进一步研究离散病毒进化、流行病学、发病机制以及预防/治疗干预提供了基础。