MOE Key Laboratory of Bioinformatics and Bioinformatics Division, TNLIST/Department of Automation, Tsinghua University, Beijing 1000084, China.
Bioinformatics. 2011 Jul 1;27(13):i167-76. doi: 10.1093/bioinformatics/btr213.
Pinpointing genes that underlie human inherited diseases among candidate genes in susceptibility genetic regions is the primary step towards the understanding of pathogenesis of diseases. Although several probabilistic models have been proposed to prioritize candidate genes using phenotype similarities and protein-protein interactions, no combinatorial approaches have been proposed in the literature.
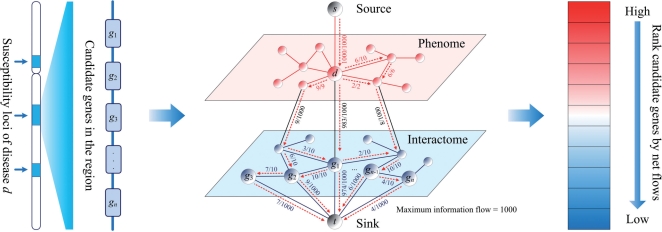
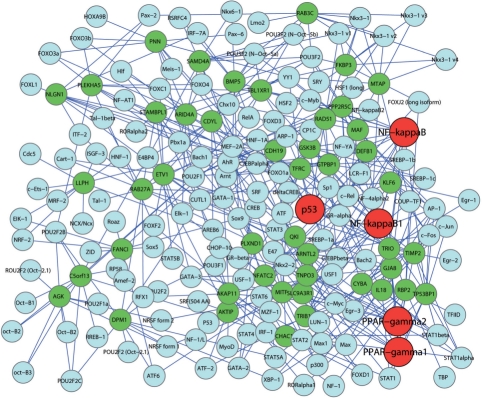
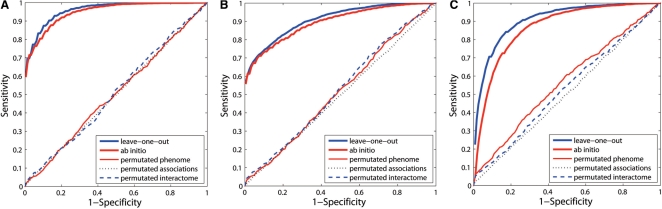
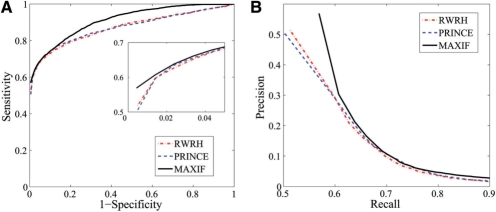
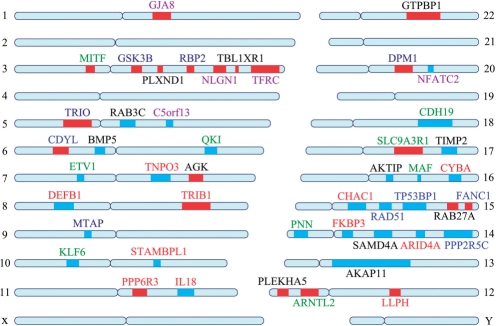
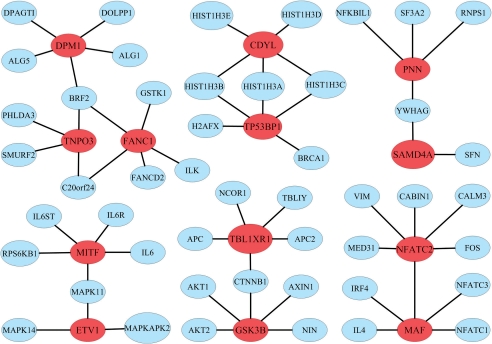
We propose the first combinatorial approach for prioritizing candidate genes. We first construct a phenome-interactome network by integrating the given phenotype similarity profile, protein-protein interaction network and associations between diseases and genes. Then, we introduce a computational method called MAXIF to maximize the information flow in this network for uncovering genes that underlie diseases. We demonstrate the effectiveness of this method in prioritizing candidate genes through a series of cross-validation experiments, and we show the possibility of using this method to identify diseases with which a query gene may be associated. We demonstrate the competitive performance of our method through a comparison with two existing state-of-the-art methods, and we analyze the robustness of our method with respect to the parameters involved. As an example application, we apply our method to predict driver genes in 50 copy number aberration regions of melanoma. Our method is not only able to identify several driver genes that have been reported in the literature, it also shed some new biological insights on the understanding of the modular property and transcriptional regulation scheme of these driver genes.
在易感遗传区域的候选基因中确定导致人类遗传疾病的基因是理解疾病发病机制的首要步骤。尽管已经提出了几种概率模型,通过表型相似性和蛋白质-蛋白质相互作用来优先考虑候选基因,但文献中尚未提出组合方法。
我们提出了第一个用于优先考虑候选基因的组合方法。我们首先通过整合给定的表型相似性谱、蛋白质-蛋白质相互作用网络以及疾病和基因之间的关联,构建一个表型-互作网络。然后,我们引入了一种称为 MAXIF 的计算方法,以最大化该网络中的信息流,从而揭示导致疾病的基因。我们通过一系列交叉验证实验证明了该方法在优先考虑候选基因方面的有效性,并展示了使用该方法识别与查询基因可能相关的疾病的可能性。我们通过与两种现有的最先进方法进行比较,展示了我们方法的竞争性能,并分析了该方法对所涉及参数的稳健性。作为一个应用示例,我们将我们的方法应用于预测黑色素瘤 50 个拷贝数缺失区域中的驱动基因。我们的方法不仅能够识别文献中已经报道的几个驱动基因,还为理解这些驱动基因的模块性质和转录调控方案提供了一些新的生物学见解。