Li Feng, Nie Lei, Wu Gang, Qiao Jianjun, Zhang Weiwen
Division of Biometrics II, Office of Biometrics/OTS/CDER/FDA, Silver Spring, MD 20993-0002, USA.
Comp Funct Genomics. 2011;2011:780973. doi: 10.1155/2011/780973. Epub 2011 May 4.
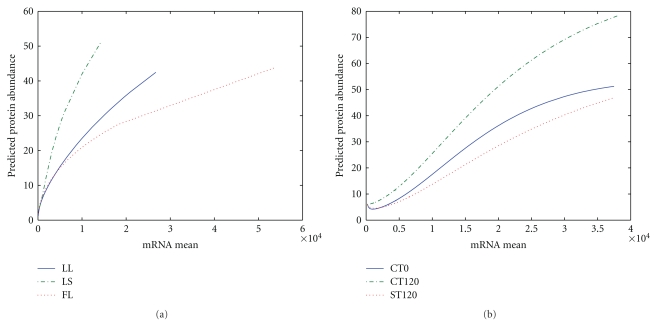
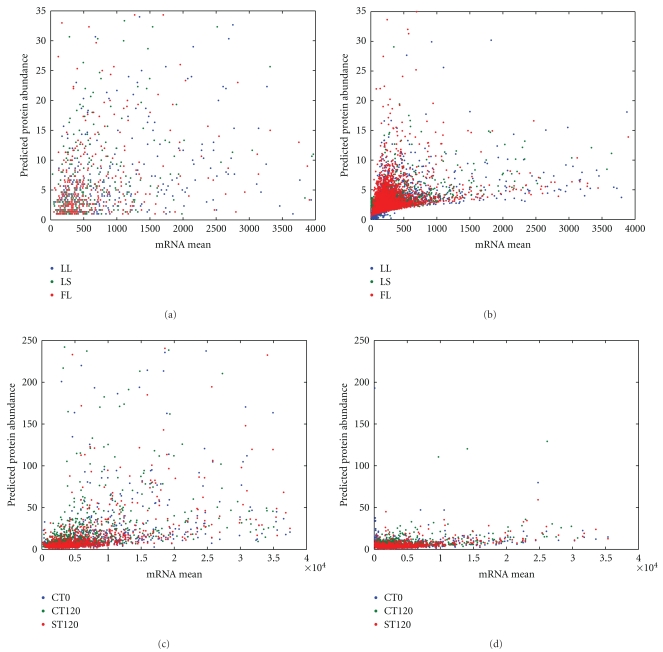
Proteomic datasets are often incomplete due to identification range and sensitivity issues. It becomes important to develop methodologies to estimate missing proteomic data, allowing better interpretation of proteomic datasets and metabolic mechanisms underlying complex biological systems. In this study, we applied an artificial neural network to approximate the relationships between cognate transcriptomic and proteomic datasets of Desulfovibrio vulgaris, and to predict protein abundance for the proteins not experimentally detected, based on several relevant predictors, such as mRNA abundance, cellular role and triple codon counts. The results showed that the coefficients of determination for the trained neural network models ranged from 0.47 to 0.68, providing better modeling than several previous regression models. The validity of the trained neural network model was evaluated using biological information (i.e. operons). To seek understanding of mechanisms causing missing proteomic data, we used a multivariate logistic regression analysis and the result suggested that some key factors, such as protein instability index, aliphatic index, mRNA abundance, effective number of codons (N(c)) and codon adaptation index (CAI) values may be ascribed to whether a given expressed protein can be detected. In addition, we demonstrated that biological interpretation can be improved by use of imputed proteomic datasets.
由于鉴定范围和灵敏度问题,蛋白质组数据集往往不完整。因此,开发估算缺失蛋白质组数据的方法变得很重要,这有助于更好地解释蛋白质组数据集以及复杂生物系统背后的代谢机制。在本研究中,我们应用人工神经网络来近似普通脱硫弧菌同源转录组和蛋白质组数据集之间的关系,并基于几个相关预测因子(如mRNA丰度、细胞功能和三联密码子计数)预测未通过实验检测到的蛋白质的丰度。结果表明,训练后的神经网络模型的决定系数在0.47至0.68之间,比之前的几个回归模型具有更好的建模效果。使用生物学信息(即操纵子)评估了训练后的神经网络模型的有效性。为了探究导致蛋白质组数据缺失的机制,我们进行了多元逻辑回归分析,结果表明,一些关键因素,如蛋白质不稳定指数、脂肪族指数、mRNA丰度、有效密码子数(N(c))和密码子适应指数(CAI)值,可能与能否检测到特定表达的蛋白质有关。此外,我们证明了使用插补后的蛋白质组数据集可以改善生物学解释。