Lin Dongdong, Zhang Jigang, Li Jingyao, Xu Chao, Deng Hong-Wen, Wang Yu-Ping
Department of Biomedical Engineering, Tulane University, New Orleans, LA, 70118, USA.
Center for Bioinformatics and Genomics, Tulane University, New Orleans, LA, 70112, USA.
BMC Bioinformatics. 2016 Jun 21;17:247. doi: 10.1186/s12859-016-1122-6.
Integrative analysis of multi-omics data is becoming increasingly important to unravel functional mechanisms of complex diseases. However, the currently available multi-omics datasets inevitably suffer from missing values due to technical limitations and various constrains in experiments. These missing values severely hinder integrative analysis of multi-omics data. Current imputation methods mainly focus on using single omics data while ignoring biological interconnections and information imbedded in multi-omics data sets.
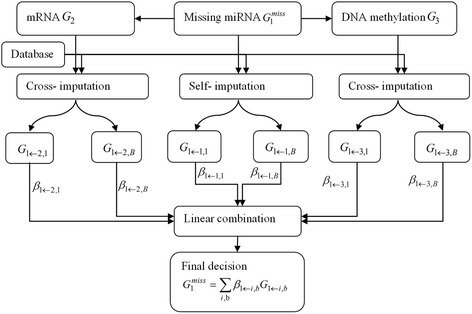
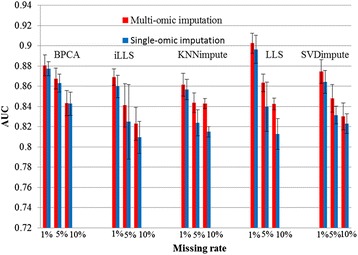
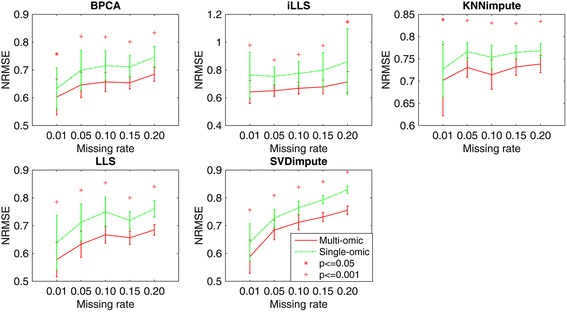
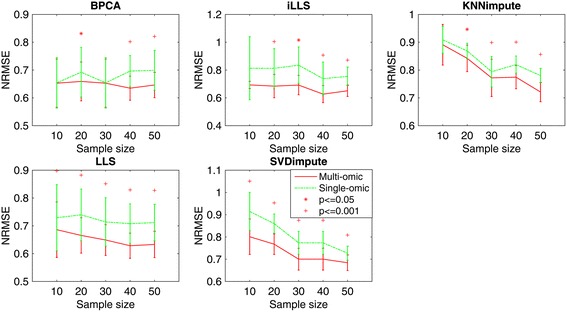
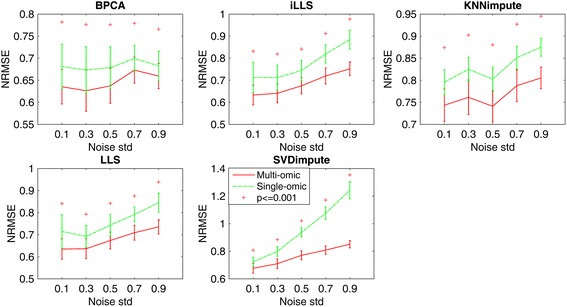
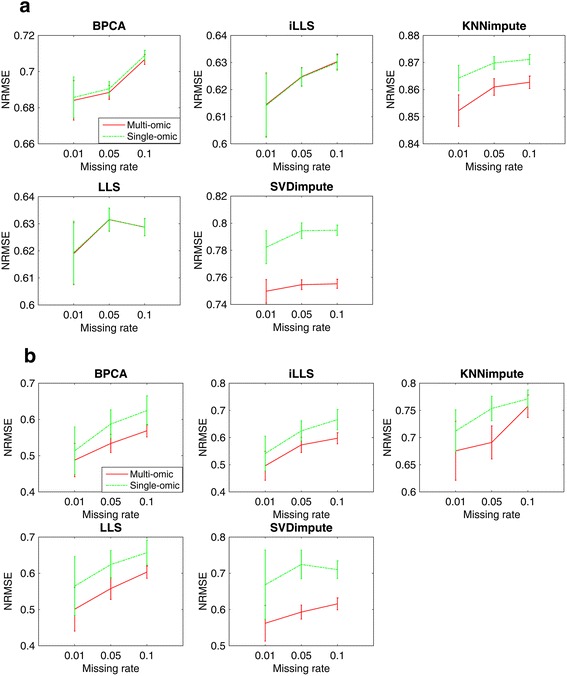
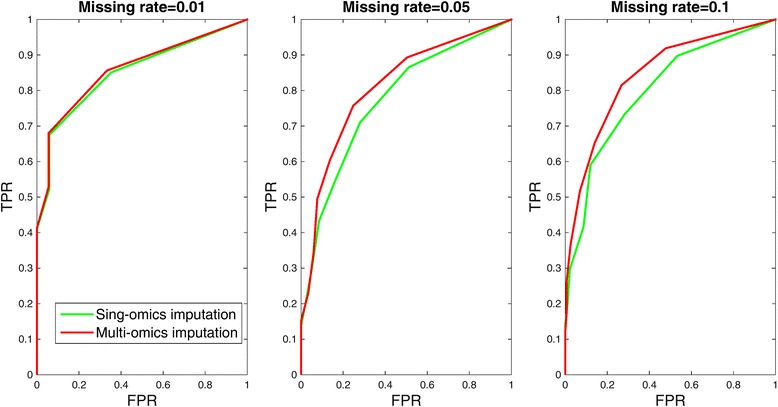
In this study, a novel multi-omics imputation method was proposed to integrate multiple correlated omics datasets for improving the imputation accuracy. Our method was designed to: 1) combine the estimates of missing value from individual omics data itself as well as from other omics, and 2) simultaneously impute multiple missing omics datasets by an iterative algorithm. We compared our method with five imputation methods using single omics data at different noise levels, sample sizes and data missing rates. The results demonstrated the advantage and efficiency of our method, consistently in terms of the imputation error and the recovery of mRNA-miRNA network structure.
We concluded that our proposed imputation method can utilize more biological information to minimize the imputation error and thus can improve the performance of downstream analysis such as genetic regulatory network construction.
多组学数据的综合分析对于揭示复杂疾病的功能机制变得越来越重要。然而,由于技术限制和实验中的各种约束,目前可用的多组学数据集不可避免地存在缺失值。这些缺失值严重阻碍了多组学数据的综合分析。当前的插补方法主要集中在使用单一组学数据,而忽略了多组学数据集中嵌入的生物联系和信息。
在本研究中,提出了一种新颖的多组学插补方法,以整合多个相关的组学数据集,提高插补准确性。我们的方法旨在:1)结合来自单个组学数据本身以及其他组学的缺失值估计,2)通过迭代算法同时插补多个缺失的组学数据集。我们在不同噪声水平、样本大小和数据缺失率下,将我们的方法与使用单一组学数据的五种插补方法进行了比较。结果在插补误差和mRNA- miRNA网络结构恢复方面一致地证明了我们方法的优势和效率。
我们得出结论,我们提出的插补方法可以利用更多的生物信息来最小化插补误差,从而可以提高下游分析(如基因调控网络构建)的性能。