State Key Laboratory of Pharmaceutical Biotechnology, School of Life Science, Nanjing University, Nanjing 210093, China.
BMC Bioinformatics. 2011 Jun 27;12:262. doi: 10.1186/1471-2105-12-262.
Current approaches for identifying transcriptional regulatory elements are mainly via the combination of two properties, the evolutionary conservation and the overrepresentation of functional elements in the promoters of co-regulated genes. Despite the development of many motif detection algorithms, the discovery of conserved motifs in a wide range of phylogenetically related promoters is still a challenge, especially for the short motifs embedded in distantly related gene promoters or very closely related promoters, or in the situation that there are not enough orthologous genes available.
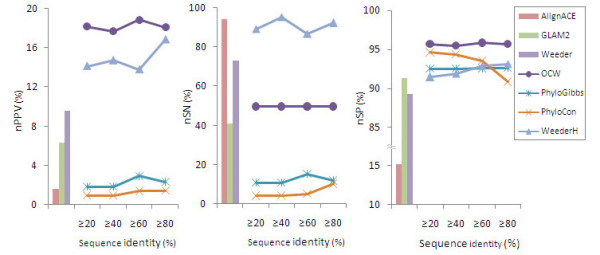
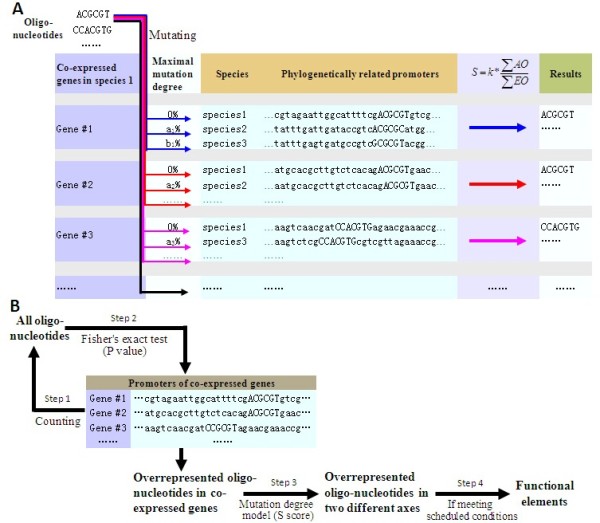
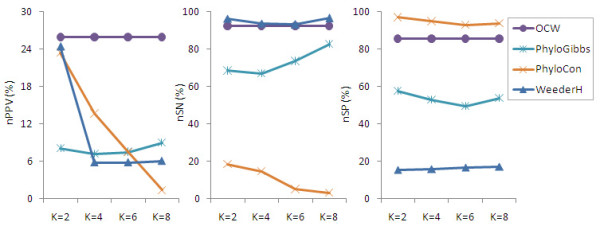
A mutation degree model is proposed and a new word counting method is developed for the identification of transcriptional regulatory elements from a set of co-expressed genes. The new method comprises two parts: 1) identifying overrepresented oligo-nucleotides in promoters of co-expressed genes, 2) estimating the conservation of the oligo-nucleotides in promoters of phylogenetically related genes by the mutation degree model. Compared with the performance of other algorithms, our method shows the advantages of low false positive rate and higher specificity, especially the robustness to noisy data. Applying the method to co-expressed gene sets from Arabidopsis, most of known cis-elements were successfully detected. The tool and example are available at http://mcube.nju.edu.cn/jwang/lab/soft/ocw/OCW.html.
The mutation degree model proposed in this paper is adapted to phylogenetic data of different qualities, and to a wide range of evolutionary distances. The new word-counting method based on this model has the advantage of better performance in detecting short sequence of cis-elements from co-expressed genes of eukaryotes and is robust to less complete phylogenetic data.
目前识别转录调控元件的方法主要是结合两个特性,即进化保守性和功能元件在共调控基因启动子中的过度表达。尽管已经开发了许多基序检测算法,但在广泛的系统发育相关启动子中发现保守基序仍然是一个挑战,特别是对于嵌入在远缘基因启动子或非常近缘基因启动子中的短基序,或者在没有足够的直系同源基因可用的情况下。
提出了一种突变程度模型,并开发了一种新的单词计数方法,用于从一组共表达基因中识别转录调控元件。该新方法包括两部分:1)识别共表达基因启动子中过度表达的寡核苷酸,2)通过突变程度模型估计系统发育相关基因启动子中寡核苷酸的保守性。与其他算法的性能相比,我们的方法具有低假阳性率和更高特异性的优势,特别是对噪声数据的稳健性。将该方法应用于拟南芥的共表达基因集,成功检测到了大多数已知的顺式元件。该工具和示例可在 http://mcube.nju.edu.cn/jwang/lab/soft/ocw/OCW.html 上获得。
本文提出的突变程度模型适用于不同质量和广泛进化距离的系统发育数据。基于该模型的新单词计数方法在检测真核生物共表达基因中短序列的顺式元件方面具有更好的性能,并且对不完整的系统发育数据具有稳健性。