Department of Life Sciences, National Cheng Kung University, Tainan 701, Taiwan.
BMC Genomics. 2011 Jul 12;12:360. doi: 10.1186/1471-2164-12-360.
Orchids are one of the most diversified angiosperms, but few genomic resources are available for these non-model plants. In addition to the ecological significance, Phalaenopsis has been considered as an economically important floriculture industry worldwide. We aimed to use massively parallel 454 pyrosequencing for a global characterization of the Phalaenopsis transcriptome.
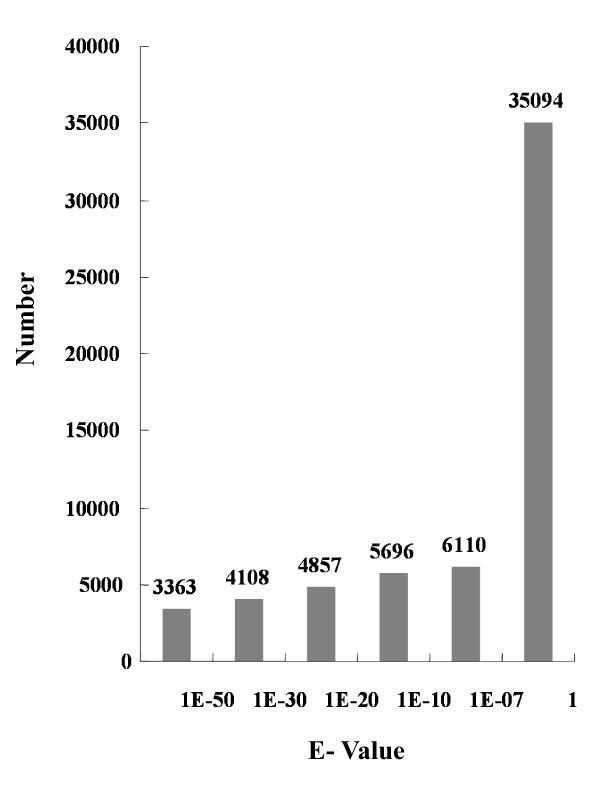
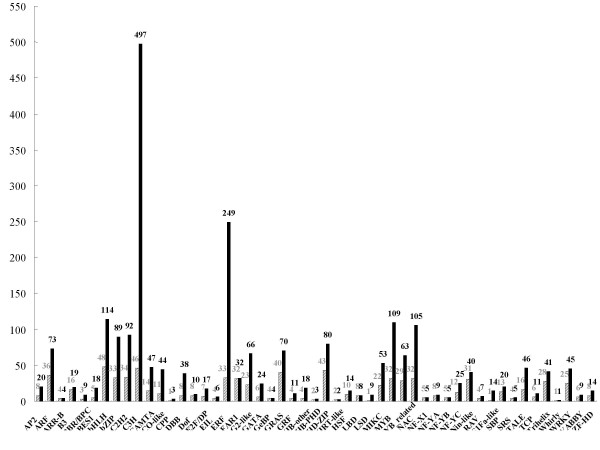
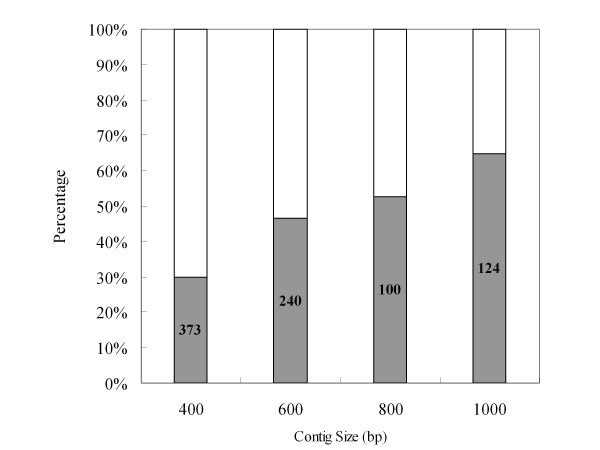
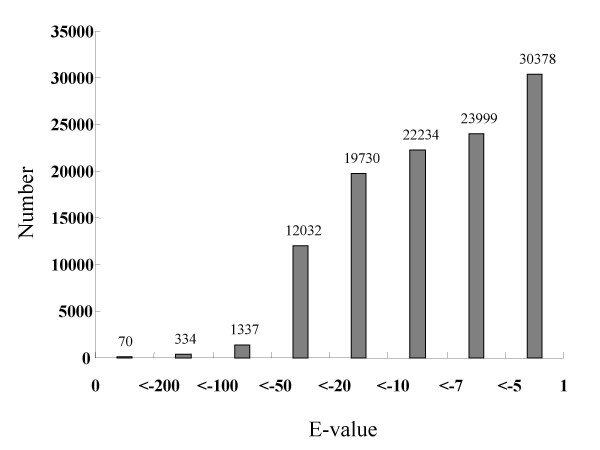
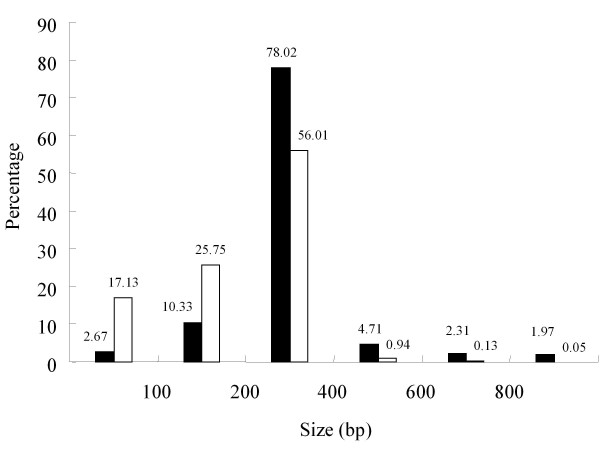
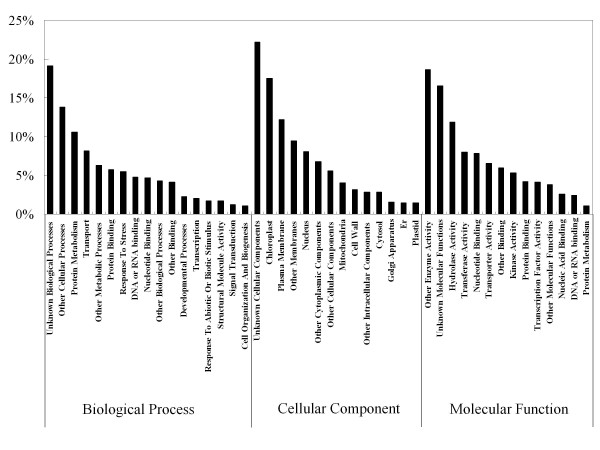
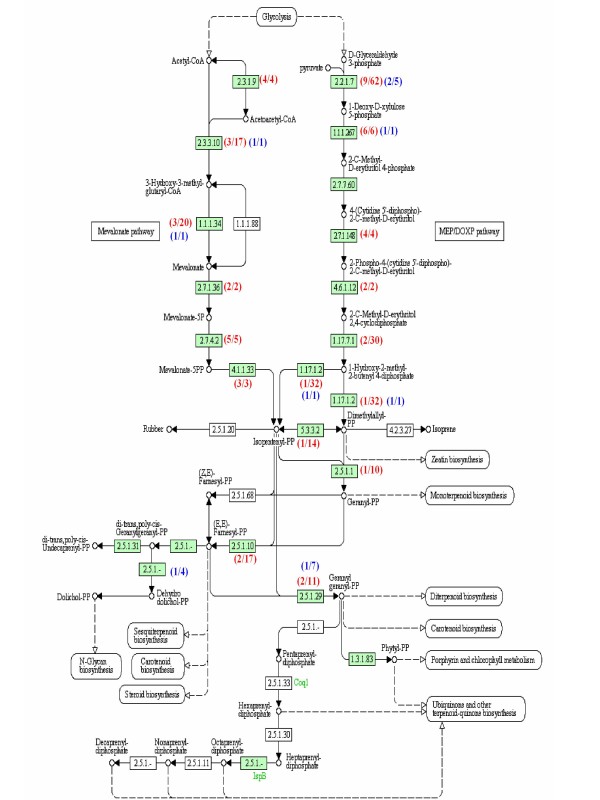
To maximize sequence diversity, we pooled RNA from 10 samples of different tissues, various developmental stages, and biotic- or abiotic-stressed plants. We obtained 206,960 expressed sequence tags (ESTs) with an average read length of 228 bp. These reads were assembled into 8,233 contigs and 34,630 singletons. The unigenes were searched against the NCBI non-redundant (NR) protein database. Based on sequence similarity with known proteins, these analyses identified 22,234 different genes (E-value cutoff, e-7). Assembled sequences were annotated with Gene Ontology, Gene Family and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Among these annotations, over 780 unigenes encoding putative transcription factors were identified.
Pyrosequencing was effective in identifying a large set of unigenes from Phalaenopsis. The informative EST dataset we developed constitutes a much-needed resource for discovery of genes involved in various biological processes in Phalaenopsis and other orchid species. These transcribed sequences will narrow the gap between study of model organisms with many genomic resources and species that are important for ecological and evolutionary studies.
兰花是被子植物中最多样化的物种之一,但这些非模式植物的基因组资源却很少。除了生态意义之外,蝴蝶兰还被认为是全球具有重要经济价值的花卉产业。我们旨在利用大规模平行 454 焦磷酸测序技术对蝴蝶兰转录组进行全面描述。
为了最大限度地增加序列多样性,我们汇集了来自不同组织、不同发育阶段以及生物或非生物胁迫植株的 10 个样本的 RNA。我们获得了 206960 条平均读长为 228bp 的表达序列标签(EST)。这些读取序列被组装成 8233 条 contigs 和 34630 条 singletons。将 unigenes 与 NCBI 非冗余(NR)蛋白质数据库进行了搜索。基于与已知蛋白质的序列相似性,这些分析鉴定了 22234 个不同的基因(E 值截止值,e-7)。组装序列用基因本体论(GO)、基因家族和京都基因与基因组百科全书(KEGG)途径进行了注释。在这些注释中,鉴定出了超过 780 个编码假定转录因子的 unigenes。
焦磷酸测序技术有效地从蝴蝶兰中鉴定出了大量的 unigenes。我们开发的信息丰富的 EST 数据集构成了发现蝴蝶兰和其他兰花物种中各种生物学过程相关基因的急需资源。这些转录序列将缩小具有大量基因组资源的模式生物研究与对生态和进化研究很重要的物种之间的差距。