Section of Molecular and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, California 90089, USA.
BMC Genomics. 2011 Jul 14;12:364. doi: 10.1186/1471-2164-12-364.
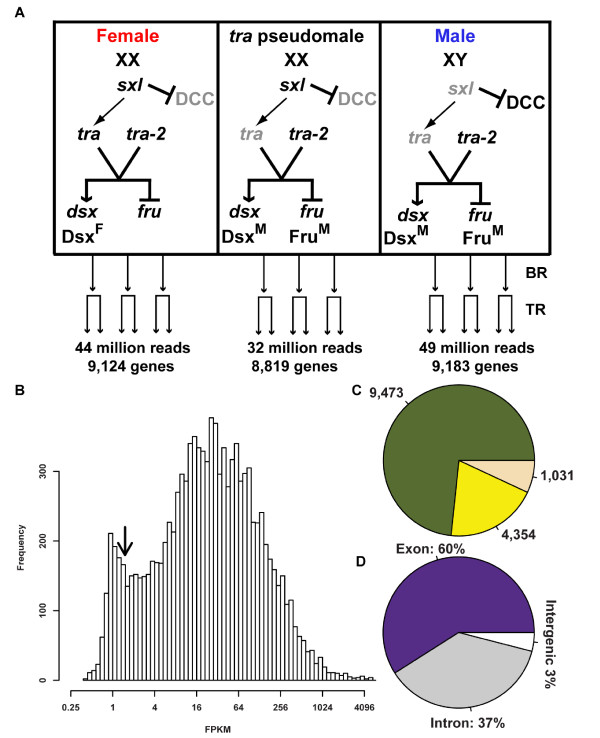
Understanding animal development and physiology at a molecular-biological level has been advanced by the ability to determine at high resolution the repertoire of mRNA molecules by whole transcriptome resequencing. This includes the ability to detect and quantify rare abundance transcripts and isoform-specific mRNA variants produced from a gene.The sex hierarchy consists of a pre-mRNA splicing cascade that directs the production of sex-specific transcription factors that specify nearly all sexual dimorphism. We have used deep RNA sequencing to gain insight into how the Drosophila sex hierarchy generates somatic sex differences, by examining gene and transcript isoform expression differences between the sexes in adult head tissues.
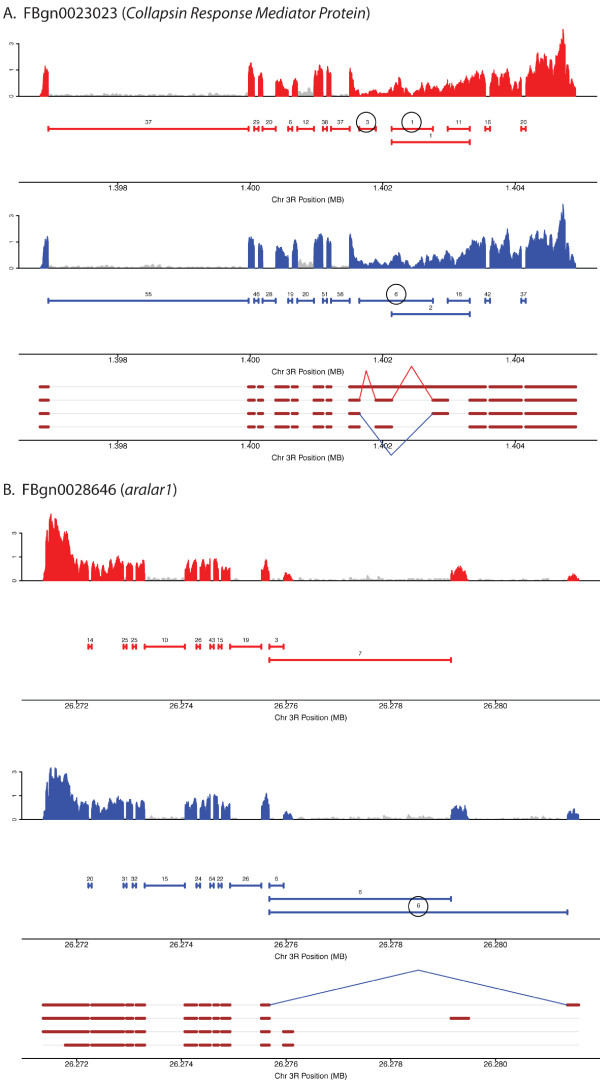
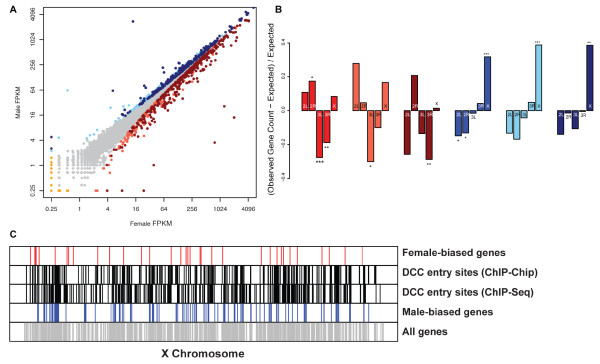
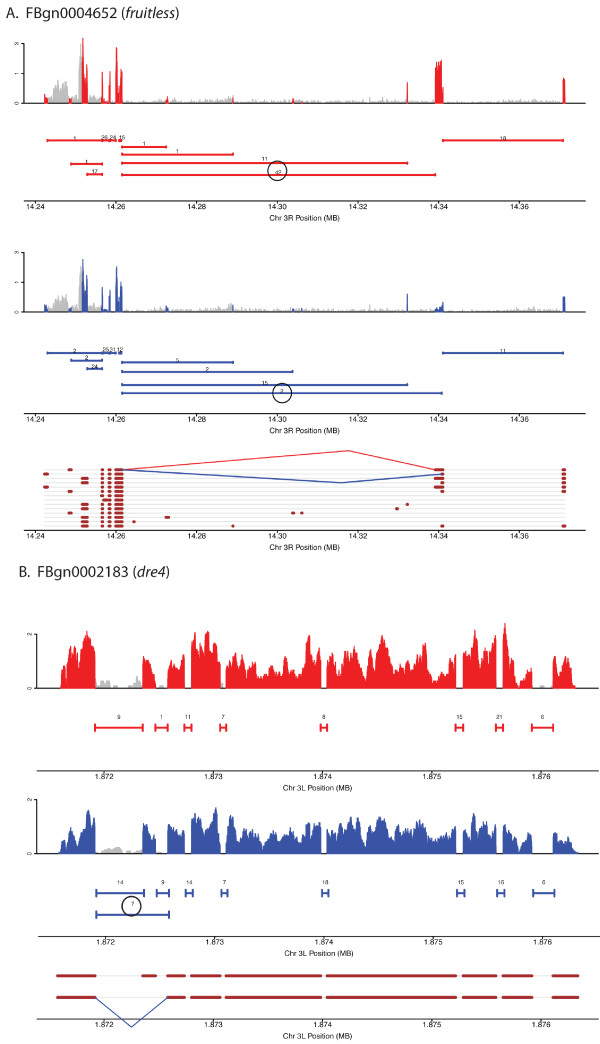
Here we find 1,381 genes that differ in overall expression levels and 1,370 isoform-specific transcripts that differ between males and females. Additionally, we find 512 genes not regulated downstream of transformer that are significantly more highly expressed in males than females. These 512 genes are enriched on the × chromosome and reside adjacent to dosage compensation complex entry sites, which taken together suggests that their residence on the × chromosome might be sufficient to confer male-biased expression. There are no transcription unit structural features, from a set of features, that are robustly significantly different in the genes with significant sex differences in the ratio of isoform-specific transcripts, as compared to random isoform-specific transcripts, suggesting that there is no single molecular mechanism that generates isoform-specific transcript differences between the sexes, even though the sex hierarchy is known to include three pre-mRNA splicing factors.
We identify thousands of genes that show sex-specific differences in overall gene expression levels, and identify hundreds of additional genes that have differences in the abundance of isoform-specific transcripts. No transcription unit structural feature was robustly enriched in the sex-differentially expressed transcript isoforms. Additionally, we found that many genes with male-biased expression were enriched on the × chromosome and reside adjacent to dosage compensation entry sites, suggesting that differences in sex chromosome composition contributes to dimorphism in gene expression. Taken together, this study provides new insight into the molecular underpinnings of sexual differentiation.
通过全转录组重测序能够高分辨率地确定 mRNA 分子谱,从而在分子生物学水平上深入了解动物的发育和生理学。这包括检测和量化稀有丰度的转录本以及从基因产生的同种型特异性 mRNA 变体的能力。性别等级由前体 mRNA 剪接级联组成,该级联指导产生特异性转录因子,特异性转录因子指定几乎所有的性别二态性。我们通过检查成年头组织中两性之间的基因和转录本同种型表达差异,利用深度 RNA 测序深入了解果蝇性别等级如何产生体细胞性别差异。
在这里,我们发现 1381 个基因在总体表达水平上存在差异,1370 个同种型特异性转录本在两性之间存在差异。此外,我们还发现 512 个不受 transformer 下游调控的基因在雄性中比雌性显著高表达。这 512 个基因在 X 染色体上富集,并位于剂量补偿复合物进入位点附近,这表明它们在 X 染色体上的位置足以赋予雄性偏倚表达。在具有显著性别差异的基因中,没有转录单元结构特征,从一组特征来看,在与随机同种型特异性转录本相比,具有显著性别差异的同种型特异性转录本的基因中,没有一个特征是显著不同的,这表明即使性别等级包括三个前体 mRNA 剪接因子,也没有单一的分子机制可以产生两性之间的同种型特异性转录本差异。
我们鉴定出数千个在整体基因表达水平上表现出性别特异性差异的基因,并鉴定出数百个具有同种型特异性转录本丰度差异的额外基因。没有转录单元结构特征在性别差异表达的转录本同种型中丰富。此外,我们发现许多雄性偏倚表达的基因在 X 染色体上富集,并位于剂量补偿进入位点附近,这表明性染色体组成的差异有助于基因表达的二态性。总之,这项研究为性分化的分子基础提供了新的见解。