Department of Biostatistics, Gillings School of Global Public Health, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Genome Biol. 2011 Jul 25;12(7):R67. doi: 10.1186/gb-2011-12-7-r67.
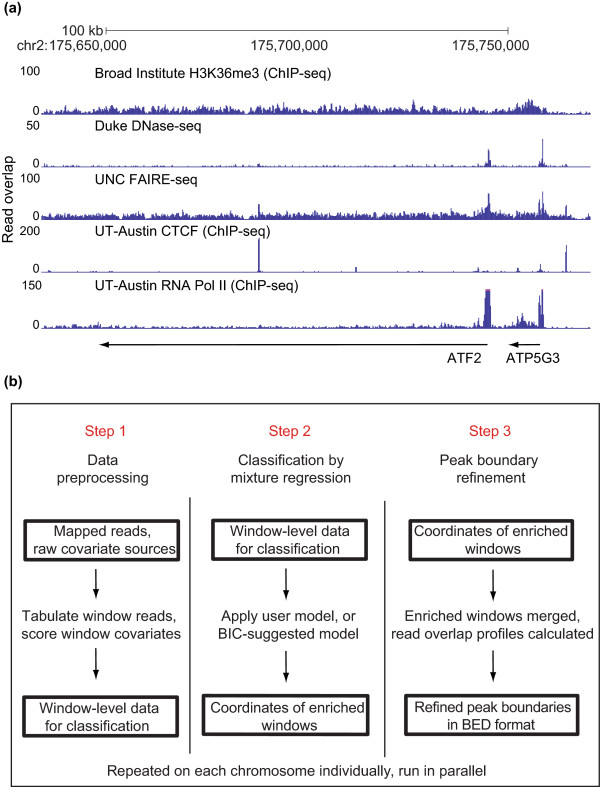
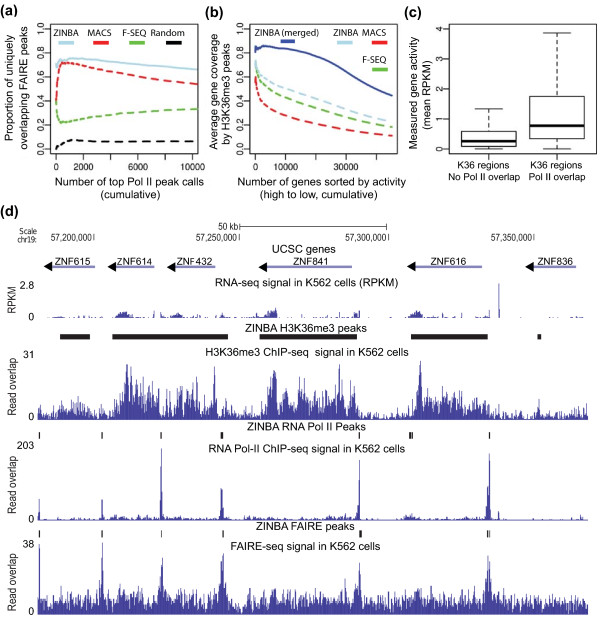
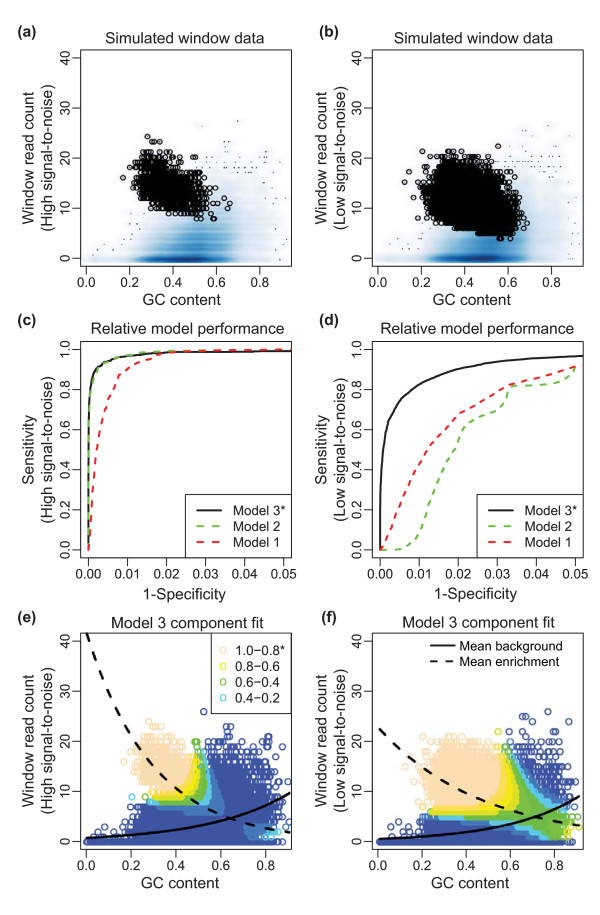
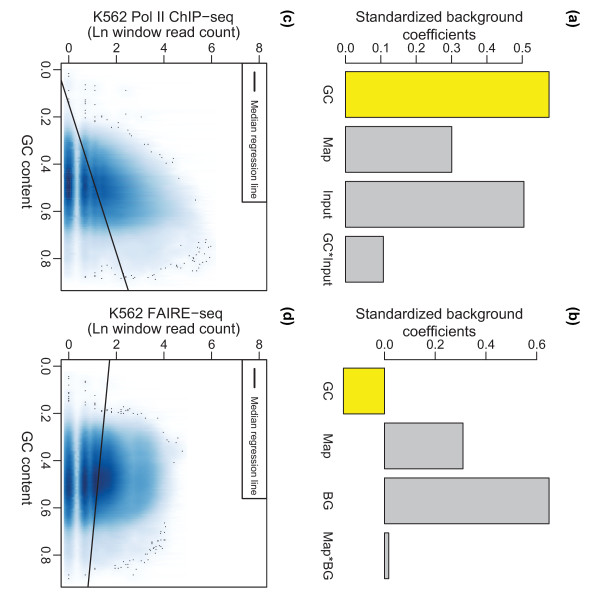
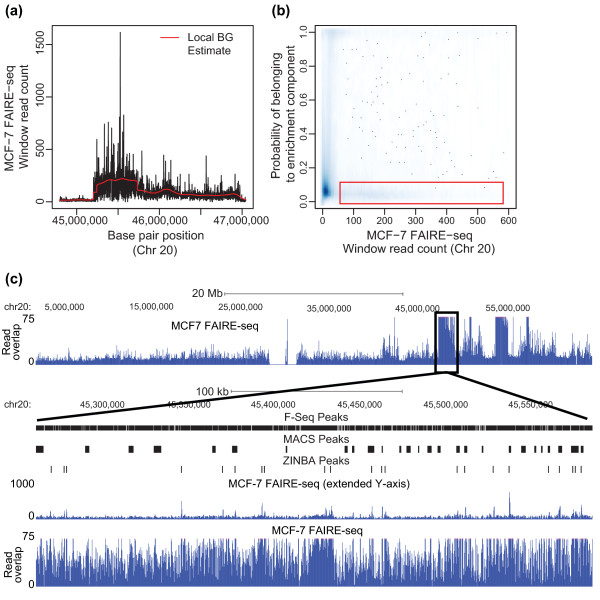
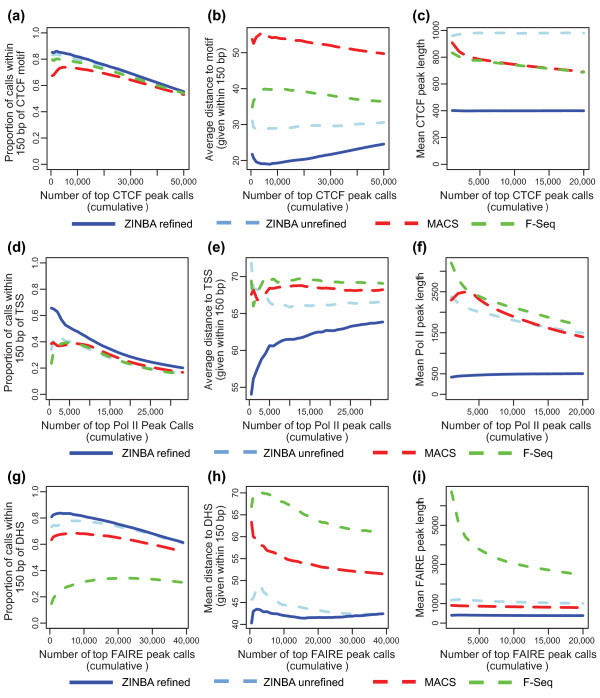
ZINBA (Zero-Inflated Negative Binomial Algorithm) identifies genomic regions enriched in a variety of ChIP-seq and related next-generation sequencing experiments (DNA-seq), calling both broad and narrow modes of enrichment across a range of signal-to-noise ratios. ZINBA models and accounts for factors that co-vary with background or experimental signal, such as G/C content, and identifies enrichment in genomes with complex local copy number variations. ZINBA provides a single unified framework for analyzing DNA-seq experiments in challenging genomic contexts.
ZINBA(零膨胀负二项式算法)可识别 ChIP-seq 和相关下一代测序实验(DNA-seq)中丰富的基因组区域,在各种信噪比范围内调用广泛和狭窄的富集模式。ZINBA 模型考虑了与背景或实验信号相关的因素,如 G/C 含量,并识别了具有复杂局部拷贝数变异的基因组中的富集。ZINBA 为分析具有挑战性基因组背景的 DNA-seq 实验提供了单一统一的框架。