Department of Pathogen Biology, Second Military Medical University, Shanghai, China.
PLoS One. 2011;6(7):e22219. doi: 10.1371/journal.pone.0022219. Epub 2011 Jul 22.
Anopheles sinensis is a competent malaria vector in China. An understanding of vector population structure is important to the vector-based malaria control programs. However, there is no adequate data of A. sinensis population genetics available yet.
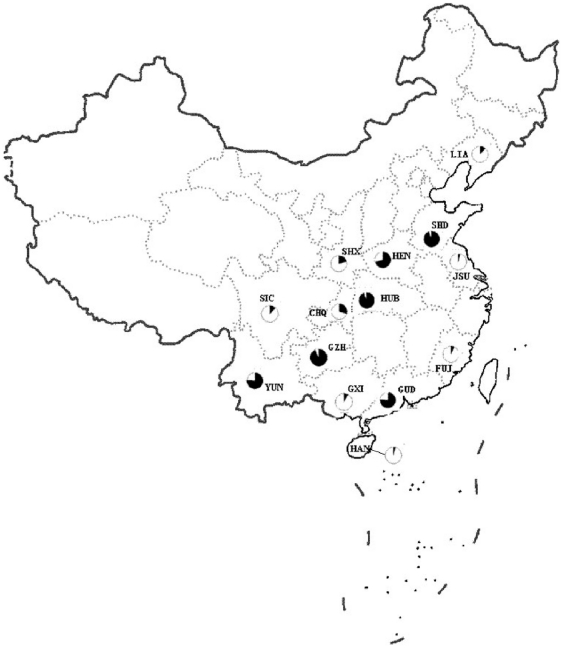
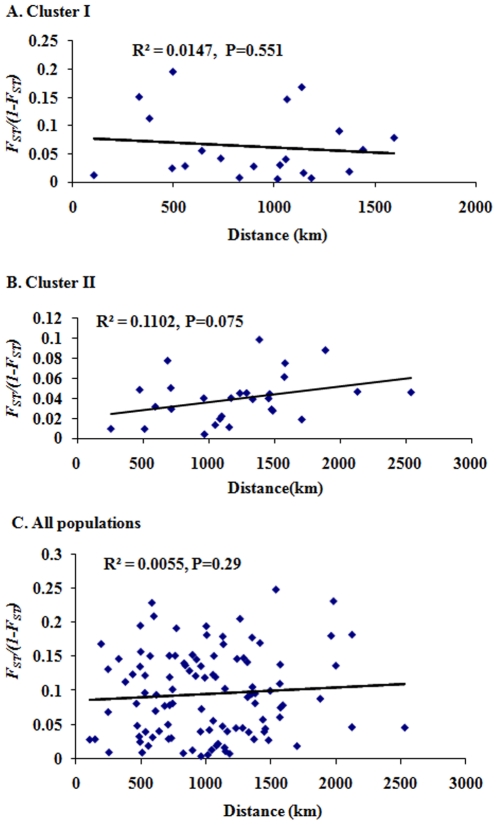
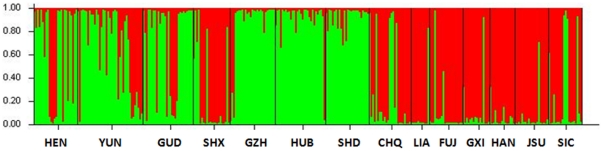
METHODOLOGY/PRINCIPAL FINDINGS: This study used 5 microsatellite loci to estimate population genetic diversity, genetic differentiation and demographic history of A. sinensis from 14 representative localities in China. All 5 microsatellite loci were highly polymorphic across populations, with high allelic richness and heterozygosity. Hardy-Weinberg disequilibrium was found in 12 populations associated with heterozygote deficits, which was likely caused by the presence of null allele and the Wahlund effect. Bayesian clustering analysis revealed two gene pools, grouping samples into two population clusters; one includes six and the other includes eight populations. Out of 14 samples, six samples were mixed with individuals from both gene pools, indicating the coexistence of two genetic units in the areas sampled. The overall differentiation between two genetic pools was moderate (F(ST) = 0.156). Pairwise differentiation between populations were lower within clusters (F(ST) = 0.008-0.028 in cluster I and F(ST) = 0.004-0.048 in cluster II) than between clusters (F(ST) = 0.120-0.201). A reduced gene flow (Nm = 1-1.7) was detected between clusters. No evidence of isolation by distance was detected among populations neither within nor between the two clusters. There are differences in effective population size (Ne = 14.3-infinite) across sampled populations.
CONCLUSIONS/SIGNIFICANCE: Two genetic pools with moderate genetic differentiation were identified in the A. sinensis populations in China. The population divergence was not correlated with geographic distance or barrier in the range. Variable effective population size and other demographic effects of historical population perturbations could be the factors affecting the population differentiation. The structured populations may limit the migration of genes under pressures/selections, such as insecticides and immune genes against malaria.
中华按蚊是中国一种具有传播疟疾能力的媒介蚊种。了解媒介种群结构对于基于媒介的疟疾控制规划非常重要。然而,目前还没有关于中华按蚊种群遗传学的充分数据。
方法/主要发现:本研究使用 5 个微卫星标记位点来估计中国 14 个代表性地区中华按蚊的种群遗传多样性、遗传分化和种群历史。所有 5 个微卫星标记位点在种群间均表现出高度多态性,具有丰富的等位基因和杂合度。在 12 个种群中发现了 Hardy-Weinberg 不平衡,这可能是由于存在无效等位基因和 Wahlund 效应所致。贝叶斯聚类分析显示存在两个基因库,将样本分为两个种群聚类;一个包含 6 个种群,另一个包含 8 个种群。在 14 个样本中,有 6 个样本与来自两个基因库的个体混合,表明在所采样的区域中存在两个遗传单位的共存。两个基因库之间的总体分化程度适中(F(ST)为 0.156)。种群间的成对分化在聚类内较低(在聚类 I 中为 F(ST) 0.008-0.028,在聚类 II 中为 F(ST) 0.004-0.048),而在聚类间较高(在聚类 I 中为 F(ST) 0.120-0.201)。在聚类间检测到较低的基因流(Nm 为 1-1.7)。在种群内和两个聚类之间都没有检测到隔离与距离的相关性。在采样的种群中,有效种群大小(Ne 为 14.3-无限大)存在差异。
结论/意义:在中国的中华按蚊种群中,确定了两个具有中等遗传分化的基因库。种群的分化与范围中的地理距离或障碍无关。历史种群扰动的有效种群大小和其他种群效应的变化可能是影响种群分化的因素。结构种群可能会限制在压力/选择下基因的迁移,如杀虫剂和针对疟疾的免疫基因。