Laboratoire de Recherche sur le Paludisme, Organisation de Coordination pour la lutte Contre les Endémies en Afrique Centrale, PO Box 288, Yaoundé, Cameroon.
Malar J. 2010 Jun 12;9:161. doi: 10.1186/1475-2875-9-161.
Anopheles nili is a widespread efficient vector of human malaria parasites in the humid savannas and forested areas of sub-Saharan Africa. Understanding An. nili population structure and gene flow patterns could be useful for the development of locally-adapted vector control measures.
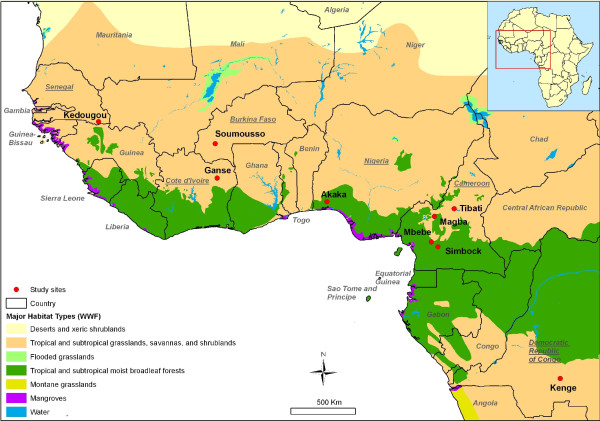
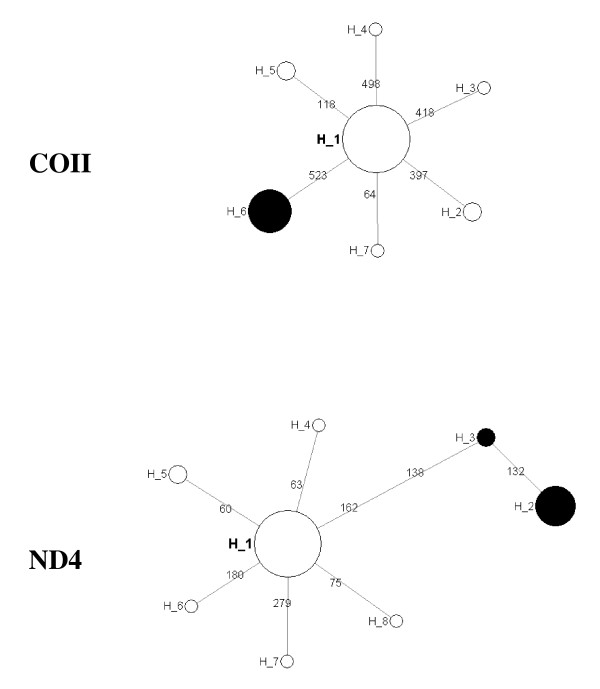
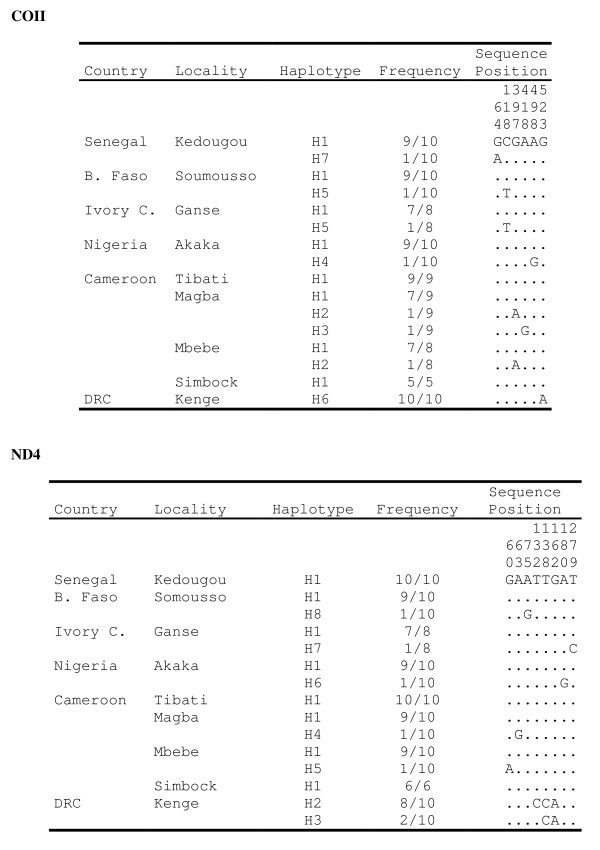
Polymorphism at eleven recently developed microsatelitte markers, and sequence variation in four genes within the 28s rDNA subunit (ITS2 and D3) and mtDNA (COII and ND4) were assessed to explore the level of genetic variability and differentiation among nine populations of An. nili from Senegal, Ivory Coast, Burkina Faso, Nigeria, Cameroon and the Democratic Republic of Congo (DRC).
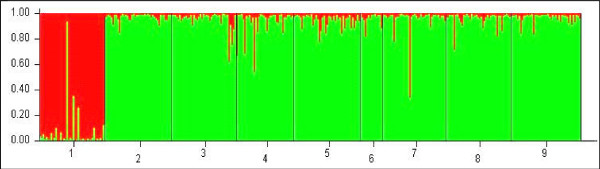
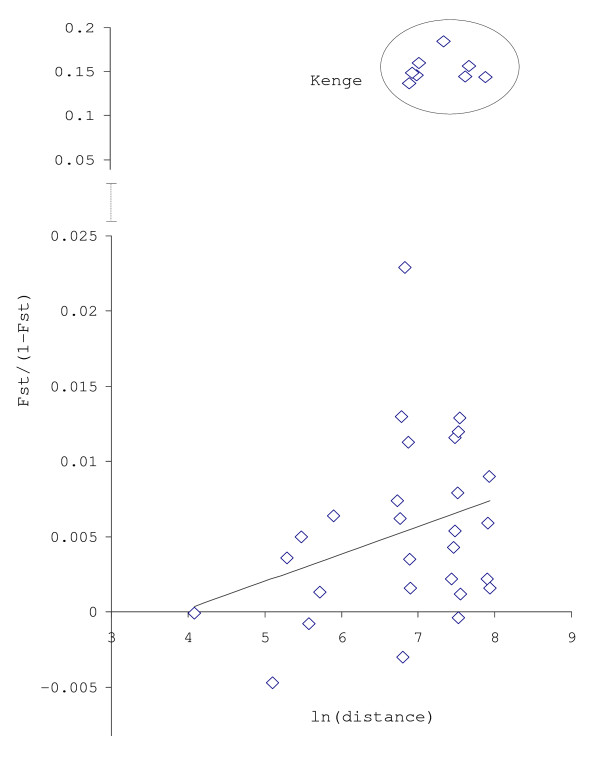
All microsatellite loci successfully amplified in all populations, showing high and very similar levels of genetic diversity in populations from West Africa and Cameroon (mean Rs = 8.10-8.88, mean He = 0.805-0.849) and much lower diversity in the Kenge population from DRC (mean Rs = 5.43, mean He = 0.594). Bayesian clustering analysis of microsatellite allelic frequencies revealed two main genetic clusters in the dataset. The first one included only the Kenge population and the second grouped together all other populations. High Fst estimates based on microsatellites (Fst > 0.118, P < 0.001) were observed in all comparisons between Kenge and all other populations. By contrast, low Fst estimates (Fst < 0.022, P < 0.05) were observed between populations within the second cluster. The correlation between genetic and geographic distances was weak and possibly obscured by demographic instability. Sequence variation in mtDNA genes matched these results, whereas low polymorphism in rDNA genes prevented detection of any population substructure at this geographical scale.
Overall, high genetic homogeneity of the An. nili gene pool was found across its distribution range in West and Central Africa, although demographic events probably resulted in a higher level of genetic isolation in the marginal population of Kenge (DRC). The role of the equatorial forest block as a barrier to gene flow and the implication of such findings for vector control are discussed.
在撒哈拉以南非洲的湿润稀树草原和森林地区,疟原虫是一种广泛存在且高效的人类疟疾传播媒介。了解按蚊尼利种群结构和基因流动模式对于制定适应当地情况的蚊虫控制措施可能很有用。
评估了来自塞内加尔、象牙海岸、布基纳法索、尼日利亚、喀麦隆和刚果民主共和国(DRC)的 9 个按蚊尼利种群的 11 个新开发的微卫星标记和 28s rDNA 亚基(ITS2 和 D3)和 mtDNA(COII 和 ND4)内的 4 个基因的序列变异,以探索遗传变异性和分化水平。
所有微卫星位点都成功地在所有种群中扩增,表明来自西非和喀麦隆的种群具有很高且非常相似的遗传多样性(平均 Rs = 8.10-8.88,平均 He = 0.805-0.849),而来自 DRC 的 Kenge 种群的多样性则要低得多(平均 Rs = 5.43,平均 He = 0.594)。微卫星等位基因频率的贝叶斯聚类分析显示,在数据集的两个主要遗传群中。第一个仅包含 Kenge 种群,第二个则将所有其他种群分组在一起。在 Kenge 与所有其他种群之间的所有比较中,都观察到基于微卫星的高 Fst 估计值(Fst > 0.118,P < 0.001)。相比之下,在第二个聚类内的种群之间观察到低 Fst 估计值(Fst < 0.022,P < 0.05)。遗传和地理距离之间的相关性较弱,并且可能被人口不稳定所掩盖。mtDNA 基因的序列变异与这些结果相匹配,而 rDNA 基因的低多态性则阻止了在该地理尺度上检测到任何种群结构。
总体而言,在安蚊尼利的分布范围内,其基因库的遗传同质性很高,尽管人口事件可能导致 Kenge (DRC)边缘种群的遗传隔离程度更高。讨论了赤道森林带作为基因流动障碍的作用以及这些发现对蚊虫控制的影响。