Medical Biotechnology Division, Center for Nanobiotechnology, School of Biosciences and Technology, Vellore Institute of Technology University, Vellore, Tamil Nadu, India.
PLoS One. 2011;6(9):e24607. doi: 10.1371/journal.pone.0024607. Epub 2011 Sep 13.
A major area of effort in current genomics is to distinguish mutations that are functionally neutral from those that contribute to disease. Single Nucleotide Polymorphisms (SNPs) are amino acid substitutions that currently account for approximately half of the known gene lesions responsible for human inherited diseases. As a result, the prediction of non-synonymous SNPs (nsSNPs) that affect protein functions and relate to disease is an important task.
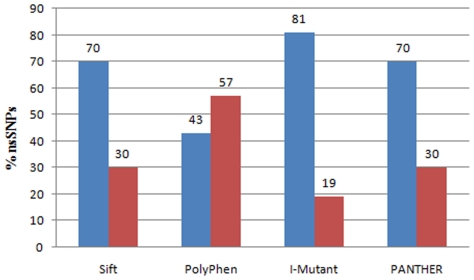
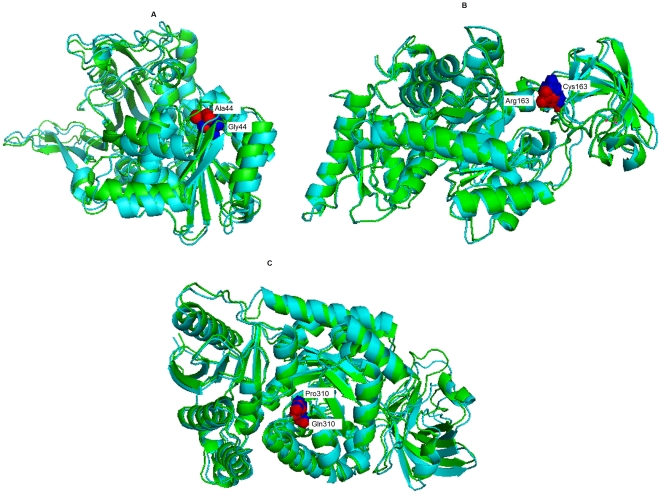
In this study, we performed a comprehensive analysis of deleterious SNPs at both functional and structural level in the respective genes associated with red blood cell metabolism disorders using bioinformatics tools. We analyzed the variants in Glucose-6-phosphate dehydrogenase (G6PD) and isoforms of Pyruvate Kinase (PKLR & PKM2) genes responsible for major red blood cell disorders. Deleterious nsSNPs were categorized based on empirical rule and support vector machine based methods to predict the impact on protein functions. Furthermore, we modeled mutant proteins and compared them with the native protein for evaluation of protein structure stability.
We argue here that bioinformatics tools can play an important role in addressing the complexity of the underlying genetic basis of Red Blood Cell disorders. Based on our investigation, we report here the potential candidate SNPs, for future studies in human Red Blood Cell disorders. Current study also demonstrates the presence of other deleterious mutations and also endorses with in vivo experimental studies. Our approach will present the application of computational tools in understanding functional variation from the perspective of structure, expression, evolution and phenotype.
当前基因组学的一个主要努力领域是区分具有功能中性的突变和导致疾病的突变。单核苷酸多态性(SNPs)是氨基酸取代,目前约占已知导致人类遗传性疾病的基因病变的一半。因此,预测影响蛋白质功能并与疾病相关的非同义 SNP(nsSNP)是一项重要任务。
在这项研究中,我们使用生物信息学工具在功能和结构水平上对与红细胞代谢紊乱相关的各自基因中的有害 SNPs 进行了全面分析。我们分析了葡萄糖-6-磷酸脱氢酶(G6PD)和丙酮酸激酶(PKLR 和 PKM2)基因的变体,这些基因负责主要的红细胞紊乱。根据经验规则和支持向量机的方法对有害 nsSNP 进行分类,以预测对蛋白质功能的影响。此外,我们对突变蛋白进行建模,并与天然蛋白进行比较,以评估蛋白质结构的稳定性。
我们认为,生物信息学工具可以在解决红细胞疾病潜在遗传基础的复杂性方面发挥重要作用。基于我们的研究,我们在此报告了潜在的候选 SNPs,供未来的人类红细胞疾病研究使用。目前的研究还表明存在其他有害突变,并与体内实验研究一致。我们的方法将从结构、表达、进化和表型的角度展示计算工具在理解功能变异方面的应用。