Mih Nathan, Brunk Elizabeth, Bordbar Aarash, Palsson Bernhard O
Bioinformatics and Systems Biology Graduate Program, University of California, San Diego, La Jolla, California, United States of America.
Department of Bioengineering, University of California, San Diego, La Jolla, California, United States of America.
PLoS Comput Biol. 2016 Jul 28;12(7):e1005039. doi: 10.1371/journal.pcbi.1005039. eCollection 2016 Jul.
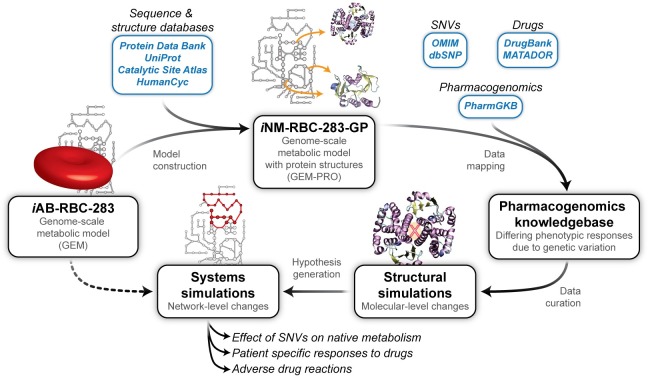
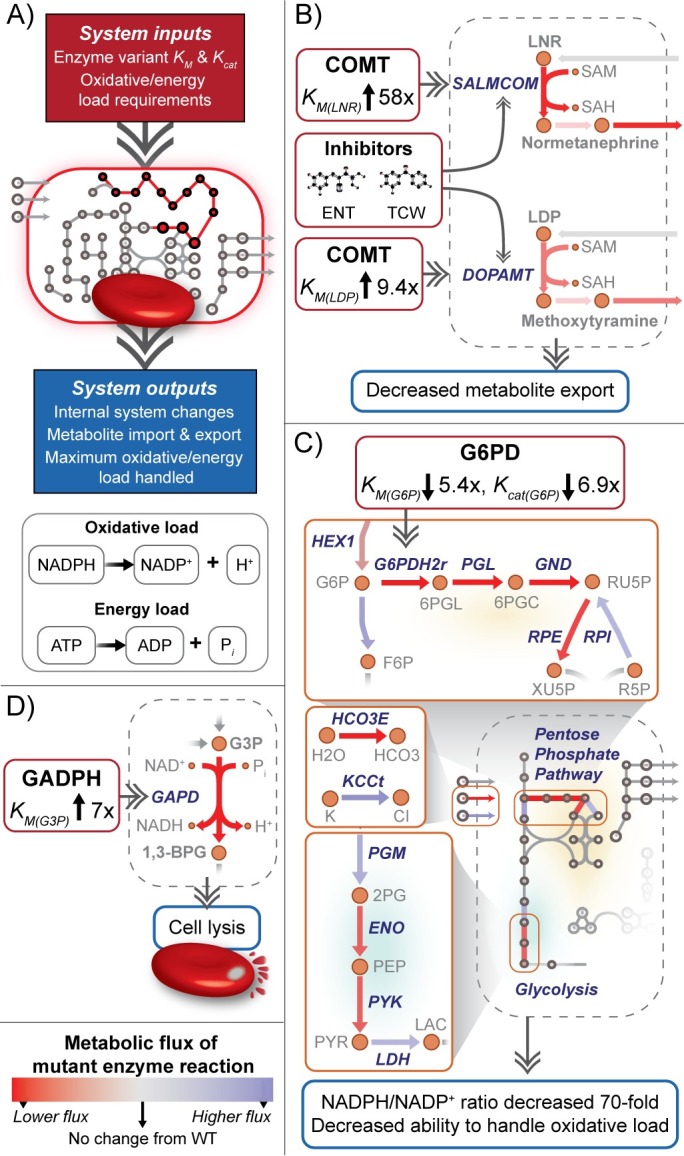
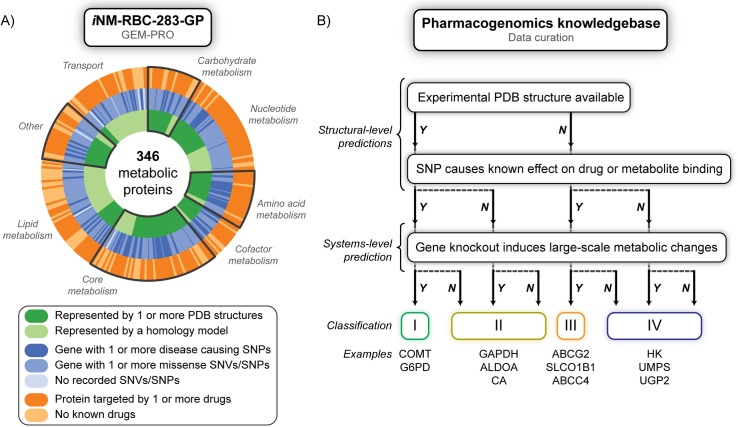
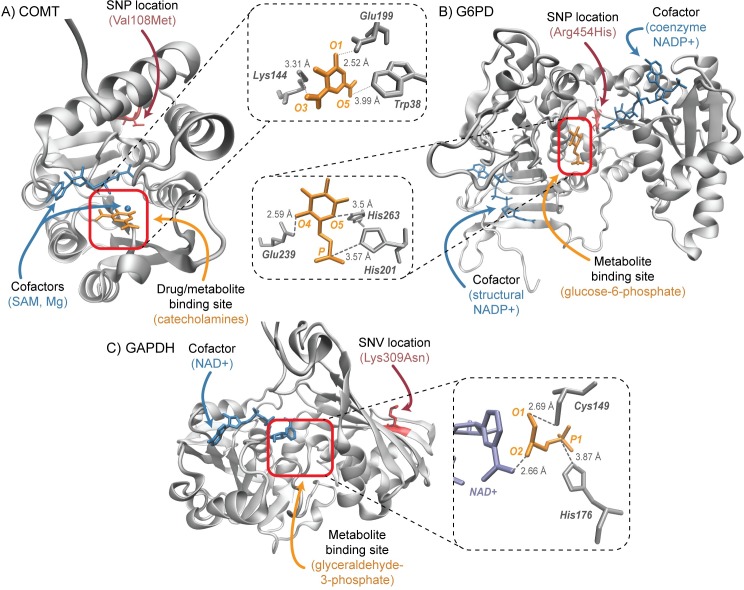
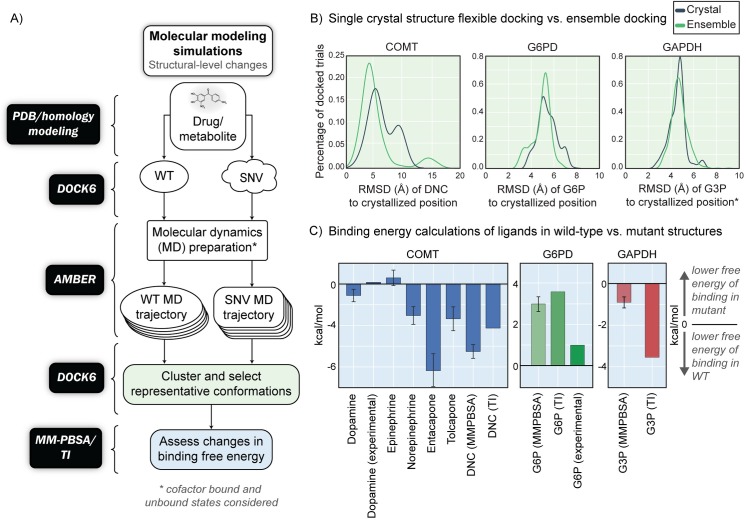
Progress in systems medicine brings promise to addressing patient heterogeneity and individualized therapies. Recently, genome-scale models of metabolism have been shown to provide insight into the mechanistic link between drug therapies and systems-level off-target effects while being expanded to explicitly include the three-dimensional structure of proteins. The integration of these molecular-level details, such as the physical, structural, and dynamical properties of proteins, notably expands the computational description of biochemical network-level properties and the possibility of understanding and predicting whole cell phenotypes. In this study, we present a multi-scale modeling framework that describes biological processes which range in scale from atomistic details to an entire metabolic network. Using this approach, we can understand how genetic variation, which impacts the structure and reactivity of a protein, influences both native and drug-induced metabolic states. As a proof-of-concept, we study three enzymes (catechol-O-methyltransferase, glucose-6-phosphate dehydrogenase, and glyceraldehyde-3-phosphate dehydrogenase) and their respective genetic variants which have clinically relevant associations. Using all-atom molecular dynamic simulations enables the sampling of long timescale conformational dynamics of the proteins (and their mutant variants) in complex with their respective native metabolites or drug molecules. We find that changes in a protein's structure due to a mutation influences protein binding affinity to metabolites and/or drug molecules, and inflicts large-scale changes in metabolism.
系统医学的进展为解决患者异质性和个性化治疗带来了希望。最近,代谢的基因组规模模型已被证明能深入了解药物治疗与系统水平脱靶效应之间的机制联系,同时被扩展到明确纳入蛋白质的三维结构。这些分子水平细节的整合,如蛋白质的物理、结构和动力学特性,显著扩展了生化网络水平特性的计算描述以及理解和预测全细胞表型的可能性。在本研究中,我们提出了一个多尺度建模框架,该框架描述了从原子细节到整个代谢网络的不同尺度的生物过程。使用这种方法,我们可以了解影响蛋白质结构和反应性的基因变异如何影响天然和药物诱导的代谢状态。作为概念验证,我们研究了三种酶(儿茶酚-O-甲基转移酶、葡萄糖-6-磷酸脱氢酶和甘油醛-3-磷酸脱氢酶)及其各自具有临床相关关联的基因变体。使用全原子分子动力学模拟能够对蛋白质(及其突变变体)与其各自的天然代谢物或药物分子复合物的长时间尺度构象动力学进行采样。我们发现,由于突变导致的蛋白质结构变化会影响蛋白质与代谢物和/或药物分子的结合亲和力,并在代谢中引起大规模变化。