Department of Bioengineering, Stanford University, Stanford, CA, USA.
BMC Bioinformatics. 2011;12 Suppl 4(Suppl 4):S3. doi: 10.1186/1471-2105-12-S4-S3. Epub 2011 Jul 5.
Single Nucleotide Polymorphisms (SNPs) are an important source of human genome variability. Non-synonymous SNPs occurring in coding regions result in single amino acid polymorphisms (SAPs) that may affect protein function and lead to pathology. Several methods attempt to estimate the impact of SAPs using different sources of information. Although sequence-based predictors have shown good performance, the quality of these predictions can be further improved by introducing new features derived from three-dimensional protein structures.
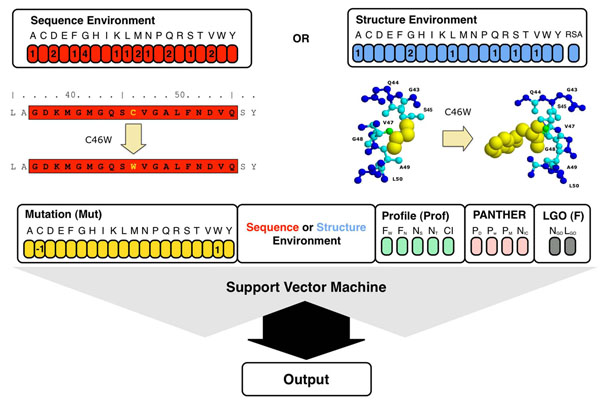
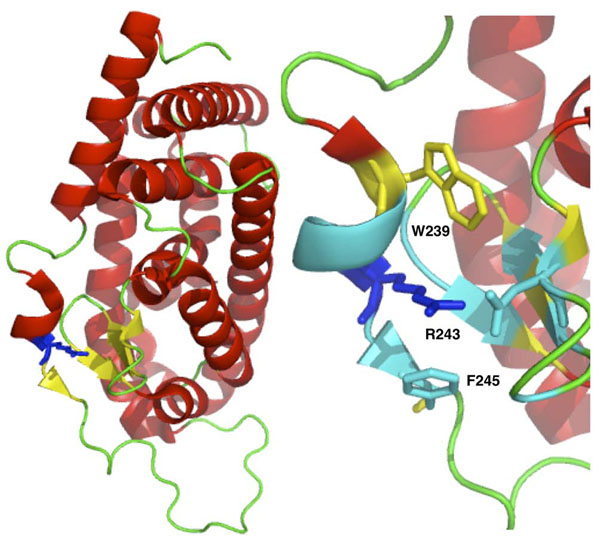
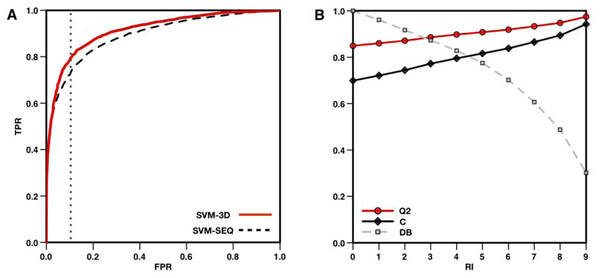
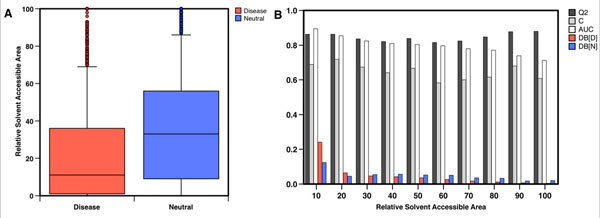
In this paper, we present a structure-based machine learning approach for predicting disease-related SAPs. We have trained a Support Vector Machine (SVM) on a set of 3,342 disease-related mutations and 1,644 neutral polymorphisms from 784 protein chains. We use SVM input features derived from the protein's sequence, structure, and function. After dataset balancing, the structure-based method (SVM-3D) reaches an overall accuracy of 85%, a correlation coefficient of 0.70, and an area under the receiving operating characteristic curve (AUC) of 0.92. When compared with a similar sequence-based predictor, SVM-3D results in an increase of the overall accuracy and AUC by 3%, and correlation coefficient by 0.06. The robustness of this improvement has been tested on different datasets and in all the cases SVM-3D performs better than previously developed methods even when compared with PolyPhen2, which explicitly considers in input protein structure information.
This work demonstrates that structural information can increase the accuracy of disease-related SAPs identification. Our results also quantify the magnitude of improvement on a large dataset. This improvement is in agreement with previously observed results, where structure information enhanced the prediction of protein stability changes upon mutation. Although the structural information contained in the Protein Data Bank is limiting the application and the performance of our structure-based method, we expect that SVM-3D will result in higher accuracy when more structural date become available.
单核苷酸多态性(SNPs)是人类基因组变异的重要来源。发生在编码区的非同义 SNPs 导致单个氨基酸多态性(SAPs),可能影响蛋白质功能并导致病理学。几种方法试图使用不同的信息来源来估计 SAP 的影响。尽管基于序列的预测器表现出良好的性能,但通过引入源自三维蛋白质结构的新特征,可以进一步提高这些预测的质量。
在本文中,我们提出了一种基于结构的机器学习方法来预测与疾病相关的 SAP。我们在 784 个蛋白质链的 3342 个疾病相关突变和 1644 个中性多态性数据集上训练了支持向量机(SVM)。我们使用源自蛋白质序列、结构和功能的 SVM 输入特征。在数据集平衡后,基于结构的方法(SVM-3D)达到了 85%的总体准确性、0.70 的相关系数和 0.92 的接收操作特征曲线下面积(AUC)。与类似的基于序列的预测器相比,SVM-3D 的总体准确性和 AUC 提高了 3%,相关系数提高了 0.06。这种改进的稳健性已在不同的数据集上进行了测试,在所有情况下,SVM-3D 的性能都优于以前开发的方法,即使与明确考虑输入蛋白质结构信息的 PolyPhen2 相比也是如此。
这项工作表明,结构信息可以提高与疾病相关的 SAP 识别的准确性。我们的结果还量化了在大型数据集上改进的幅度。这种改进与先前观察到的结果一致,其中结构信息增强了突变后蛋白质稳定性变化的预测。尽管蛋白质数据库中包含的结构信息限制了我们基于结构的方法的应用和性能,但我们预计,当更多的结构数据可用时,SVM-3D 将产生更高的准确性。