MRC Centre for Neurodegeneration Research, Institute of Psychiatry P 041, London SE5 8AF, UK.
Brain. 2011 Dec;134(Pt 12):3454-7. doi: 10.1093/brain/awr248. Epub 2011 Sep 20.
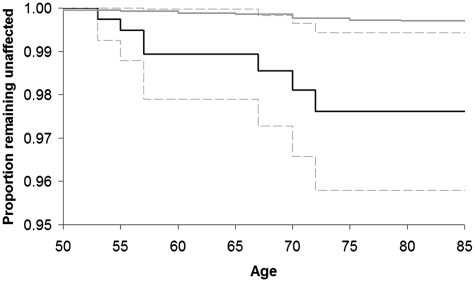
Amyotrophic lateral sclerosis is a neurodegenerative disease of motor neurons with a median survival of 2 years. Most patients have no family history of amyotrophic lateral sclerosis, but current understanding of such diseases suggests there should be an increased risk to relatives. Furthermore, it is a common question to be asked by patients and relatives in clinic. We therefore set out to determine the risk of amyotrophic lateral sclerosis to first degree relatives of patients with sporadic amyotrophic lateral sclerosis attending a specialist clinic. Case records of patients with sporadic amyotrophic lateral sclerosis seen at a tertiary referral centre over a 16-year period were reviewed, and pedigree structures extracted. All individuals who had originally presented with sporadic amyotrophic lateral sclerosis, but who subsequently had an affected first degree relative, were identified. Calculations were age-adjusted using clinic population demographics. Probands (n = 1502), full siblings (n = 1622) and full offspring (n = 1545) were identified. Eight of the siblings and 18 offspring had developed amyotrophic lateral sclerosis. The unadjusted risk of amyotrophic lateral sclerosis over the observation period was 0.5% for siblings and 1.0% for offspring. Age information was available for 476 siblings and 824 offspring. For this subset, the crude incidence of amyotrophic lateral sclerosis was 0.11% per year (0.05-0.21%) in siblings and 0.11% per year (0.06-0.19%) in offspring, and the clinic age-adjusted incidence rate was 0.12% per year (0.04-0.21%) in siblings. By age 85, siblings were found to have an 8-fold increased risk of amyotrophic lateral sclerosis, in comparison to the background population. In practice, this means the risk of remaining unaffected by age 85 dropped from 99.7% to 97.6%. Relatives of people with sporadic amyotrophic lateral sclerosis have a small but definite increased risk of being affected.
肌萎缩侧索硬化症是一种运动神经元退行性疾病,中位生存期为 2 年。大多数患者没有肌萎缩侧索硬化症的家族史,但目前对这类疾病的认识表明,亲属的风险会增加。此外,这也是临床中患者和亲属经常问到的问题。因此,我们着手确定在专门诊所就诊的散发性肌萎缩侧索硬化症患者的一级亲属患肌萎缩侧索硬化症的风险。
回顾了 16 年来在三级转诊中心就诊的散发性肌萎缩侧索硬化症患者的病历,并提取了家族结构。确定了最初表现为散发性肌萎缩侧索硬化症但随后有一级亲属受累的所有个体。使用诊所人群的人口统计学数据对计算结果进行了年龄调整。确定了先证者(n=1502)、全同胞(n=1622)和全子女(n=1545)。有 8 名同胞和 18 名子女发展为肌萎缩侧索硬化症。在观察期间,未调整的同胞肌萎缩侧索硬化症风险为 0.5%,子女为 1.0%。有 476 名同胞和 824 名子女的年龄信息可用。对于这部分人群,肌萎缩侧索硬化症的粗发病率为每年 0.11%(0.05-0.21%)在同胞中,每年 0.11%(0.06-0.19%)在子女中,诊所年龄调整后的发病率为每年 0.12%(0.04-0.21%)在同胞中。到 85 岁时,与普通人群相比,同胞患肌萎缩侧索硬化症的风险增加了 8 倍。实际上,这意味着到 85 岁时仍未受影响的风险从 99.7%下降到 97.6%。散发性肌萎缩侧索硬化症患者的亲属有患该病的小但确定的风险增加。