Institute for Systems Biology, Seattle, Washington 98109, USA.
Mol Cell Proteomics. 2012 Apr;11(4):R111.015040. doi: 10.1074/mcp.R111.015040. Epub 2011 Dec 12.
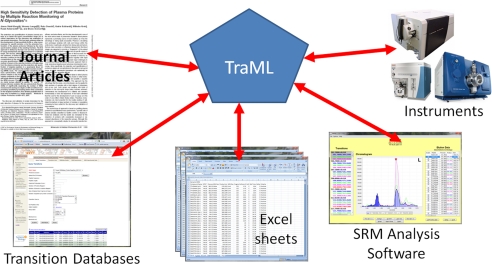
Targeted proteomics via selected reaction monitoring is a powerful mass spectrometric technique affording higher dynamic range, increased specificity and lower limits of detection than other shotgun mass spectrometry methods when applied to proteome analyses. However, it involves selective measurement of predetermined analytes, which requires more preparation in the form of selecting appropriate signatures for the proteins and peptides that are to be targeted. There is a growing number of software programs and resources for selecting optimal transitions and the instrument settings used for the detection and quantification of the targeted peptides, but the exchange of this information is hindered by a lack of a standard format. We have developed a new standardized format, called TraML, for encoding transition lists and associated metadata. In addition to introducing the TraML format, we demonstrate several implementations across the community, and provide semantic validators, extensive documentation, and multiple example instances to demonstrate correctly written documents. Widespread use of TraML will facilitate the exchange of transitions, reduce time spent handling incompatible list formats, increase the reusability of previously optimized transitions, and thus accelerate the widespread adoption of targeted proteomics via selected reaction monitoring.
靶向蛋白质组学通过选择反应监测是一种强大的质谱技术,与其他 shotgun 质谱方法相比,当应用于蛋白质组分析时,它具有更高的动态范围、更高的特异性和更低的检测限。然而,它涉及到对预定分析物的选择性测量,这需要更多的准备工作,例如为要靶向的蛋白质和肽选择适当的特征。现在有越来越多的软件程序和资源可用于选择最佳的转换以及用于检测和定量靶向肽的仪器设置,但由于缺乏标准格式,这种信息的交换受到阻碍。我们开发了一种新的标准化格式,称为 TraML,用于编码转换列表和相关元数据。除了介绍 TraML 格式外,我们还在整个社区中展示了几个实现,并提供语义验证器、广泛的文档和多个示例实例,以演示正确编写的文档。TraML 的广泛使用将促进转换的交换,减少处理不兼容列表格式所花费的时间,增加以前优化的转换的可重用性,从而加速通过选择反应监测进行靶向蛋白质组学的广泛采用。