Navarro Pedro, Kuharev Jörg, Gillet Ludovic C, Bernhardt Oliver M, MacLean Brendan, Röst Hannes L, Tate Stephen A, Tsou Chih-Chiang, Reiter Lukas, Distler Ute, Rosenberger George, Perez-Riverol Yasset, Nesvizhskii Alexey I, Aebersold Ruedi, Tenzer Stefan
Institute for Immunology, University Medical Center of the Johannes-Gutenberg University Mainz, Mainz, Germany.
Department of Biology, Institute of Molecular Systems Biology, Eidgenoessische Technische Hochschule (IMSB-ETH) Zurich, Zurich, Switzerland.
Nat Biotechnol. 2016 Nov;34(11):1130-1136. doi: 10.1038/nbt.3685. Epub 2016 Oct 3.
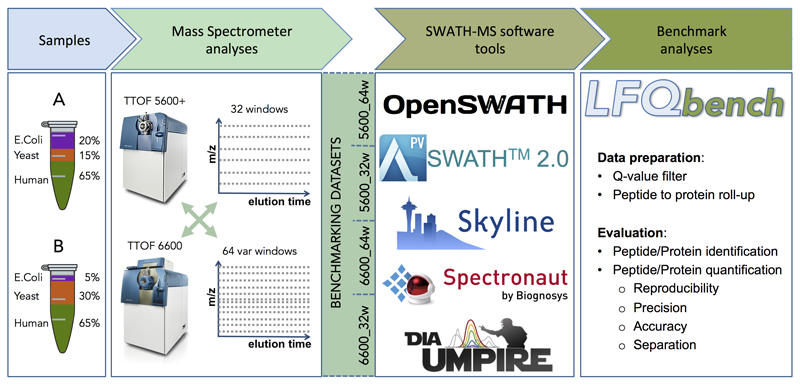
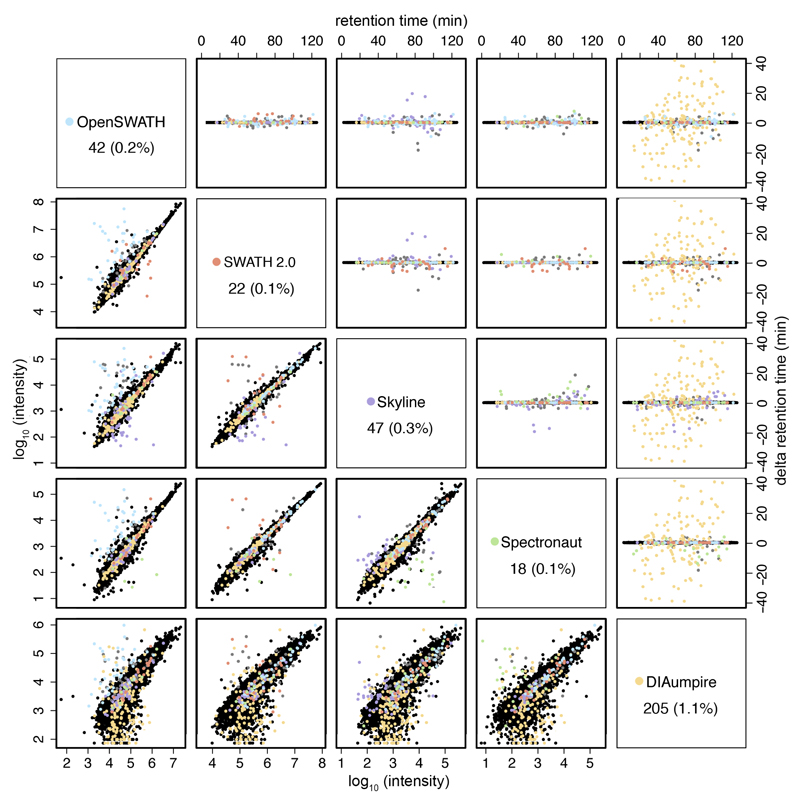
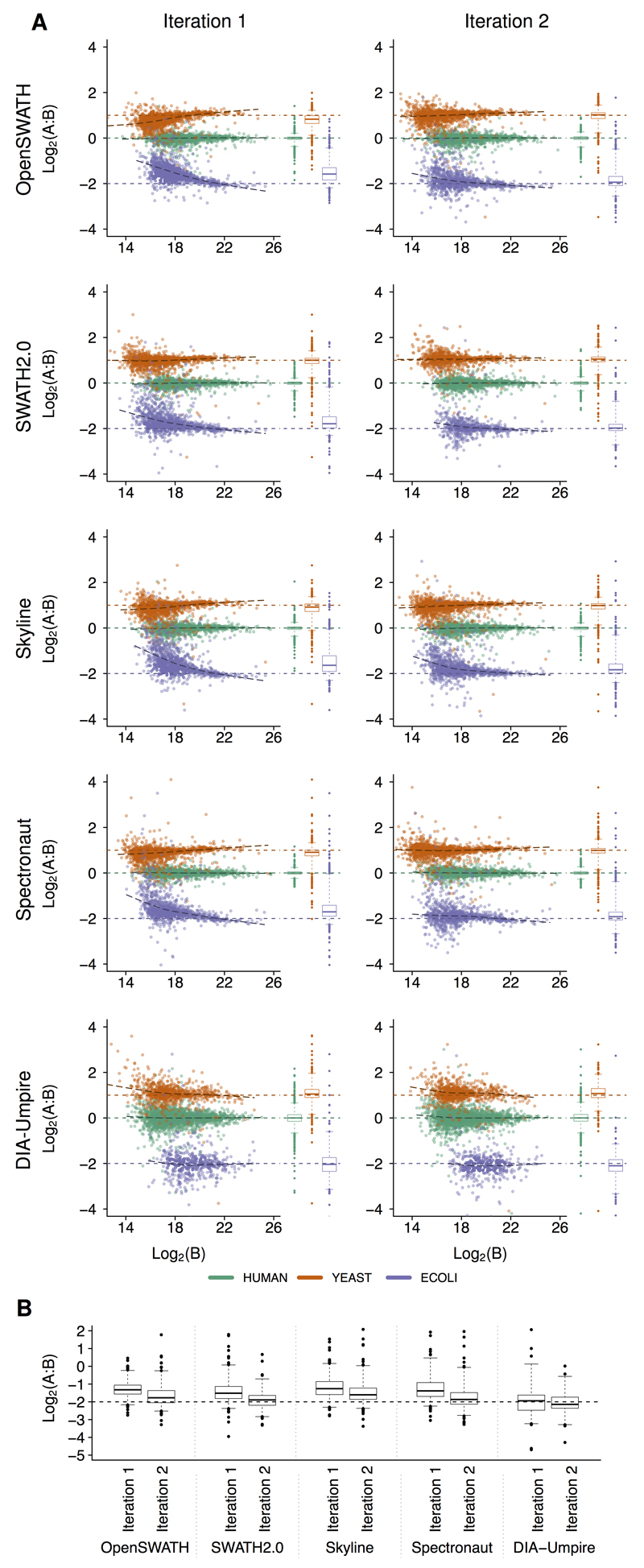
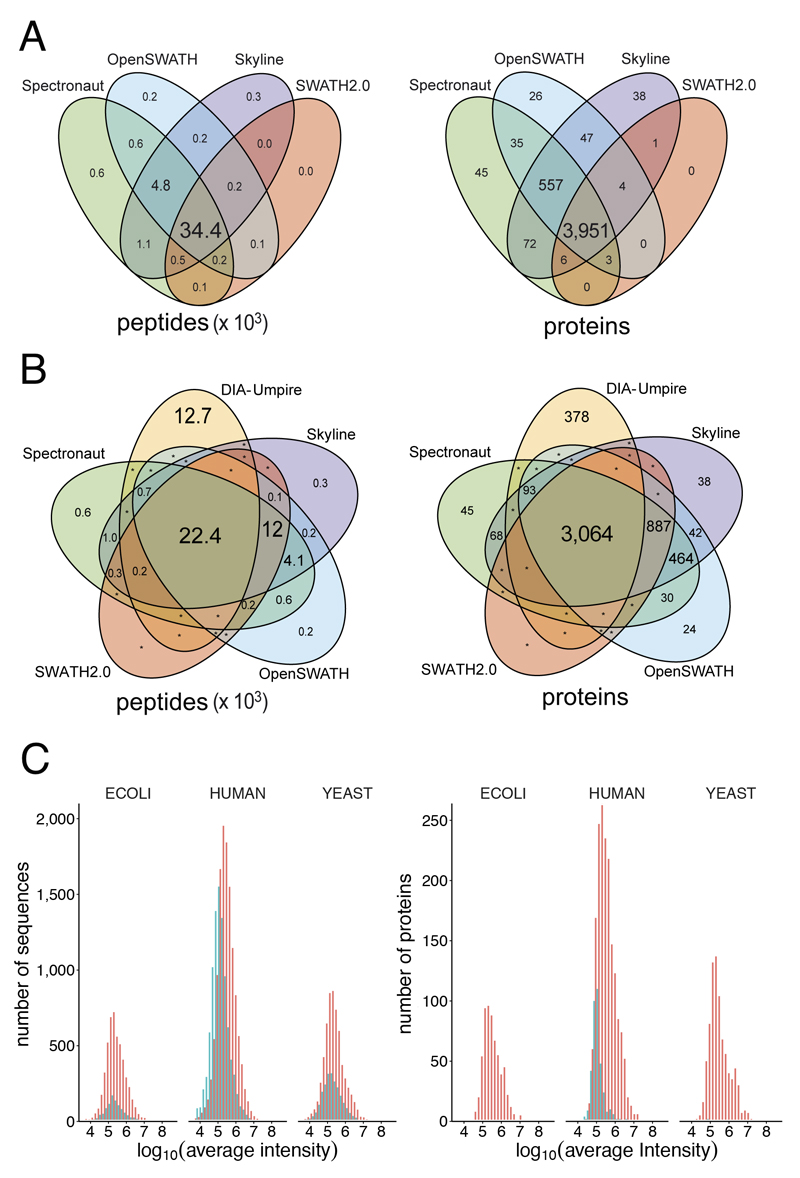
Consistent and accurate quantification of proteins by mass spectrometry (MS)-based proteomics depends on the performance of instruments, acquisition methods and data analysis software. In collaboration with the software developers, we evaluated OpenSWATH, SWATH 2.0, Skyline, Spectronaut and DIA-Umpire, five of the most widely used software methods for processing data from sequential window acquisition of all theoretical fragment-ion spectra (SWATH)-MS, which uses data-independent acquisition (DIA) for label-free protein quantification. We analyzed high-complexity test data sets from hybrid proteome samples of defined quantitative composition acquired on two different MS instruments using different SWATH isolation-window setups. For consistent evaluation, we developed LFQbench, an R package, to calculate metrics of precision and accuracy in label-free quantitative MS and report the identification performance, robustness and specificity of each software tool. Our reference data sets enabled developers to improve their software tools. After optimization, all tools provided highly convergent identification and reliable quantification performance, underscoring their robustness for label-free quantitative proteomics.
基于质谱(MS)的蛋白质组学对蛋白质进行一致且准确的定量,取决于仪器性能、采集方法和数据分析软件。我们与软件开发人员合作,评估了OpenSWATH、SWATH 2.0、Skyline、Spectronaut和DIA-Umpire这五种最广泛使用的软件方法,它们用于处理来自全理论碎片离子光谱顺序窗口采集(SWATH)-MS的数据,该技术使用数据非依赖采集(DIA)进行无标记蛋白质定量。我们分析了在两种不同的MS仪器上使用不同的SWATH隔离窗口设置从具有确定定量组成的混合蛋白质组样品中获取的高复杂性测试数据集。为了进行一致的评估,我们开发了R包LFQbench,以计算无标记定量MS中的精密度和准确度指标,并报告每个软件工具的鉴定性能、稳健性和特异性。我们的参考数据集使开发人员能够改进他们的软件工具。经过优化后,所有工具都提供了高度一致的鉴定结果和可靠的定量性能,突出了它们在无标记定量蛋白质组学方面的稳健性。