Department of Biotechnology, Indian Institute of Technology Madras, Chennai 600 036, Tamilnadu, India.
Proteome Sci. 2011 Oct 14;9 Suppl 1(Suppl 1):S13. doi: 10.1186/1477-5956-9-S1-S13.
Protein-protein interactions are important for several cellular processes. Understanding the mechanism of protein-protein recognition and predicting the binding sites in protein-protein complexes are long standing goals in molecular and computational biology.
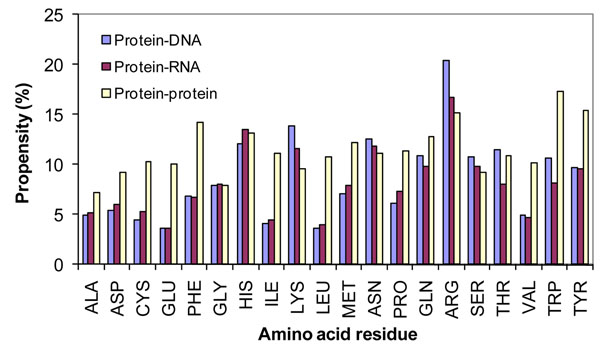
We have developed an energy based approach for identifying the binding site residues in protein-protein complexes. The binding site residues have been analyzed with sequence and structure based parameters such as binding propensity, neighboring residues in the vicinity of binding sites, conservation score and conformational switching.
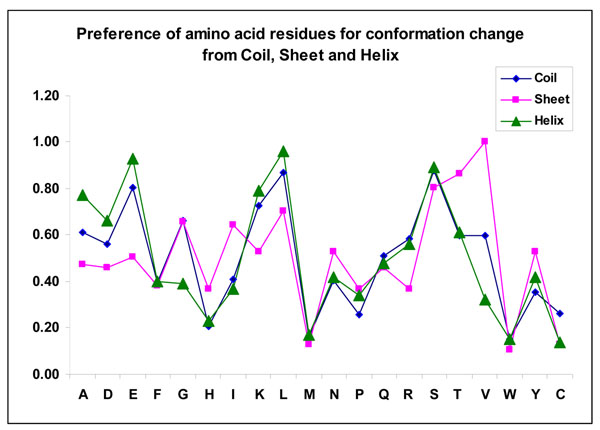
We observed that the binding propensities of amino acid residues are specific for protein-protein complexes. Further, typical dipeptides and tripeptides showed high preference for binding, which is unique to protein-protein complexes. Most of the binding site residues are highly conserved among homologous sequences. Our analysis showed that 7% of residues changed their conformations upon protein-protein complex formation and it is 9.2% and 6.6% in the binding and non-binding sites, respectively. Specifically, the residues Glu, Lys, Leu and Ser changed their conformation from coil to helix/strand and from helix to coil/strand. Leu, Ser, Thr and Val prefer to change their conformation from strand to coil/helix.
The results obtained in this study will be helpful for understanding and predicting the binding sites in protein-protein complexes.
蛋白质-蛋白质相互作用对于许多细胞过程至关重要。理解蛋白质-蛋白质识别的机制并预测蛋白质-蛋白质复合物中的结合位点是分子和计算生物学中的长期目标。
我们开发了一种基于能量的方法来识别蛋白质-蛋白质复合物中的结合位点残基。使用基于序列和结构的参数(如结合倾向、结合位点附近的邻近残基、保守得分和构象转换)分析了结合位点残基。
我们观察到氨基酸残基的结合倾向是蛋白质-蛋白质复合物特有的。此外,典型的二肽和三肽对结合具有很高的偏好性,这是蛋白质-蛋白质复合物所特有的。大多数结合位点残基在同源序列中高度保守。我们的分析表明,在蛋白质-蛋白质复合物形成时,7%的残基改变了它们的构象,而在结合和非结合位点中,分别有 9.2%和 6.6%的残基发生了改变。具体来说,残基 Glu、Lys、Leu 和 Ser 从螺旋/链构象转变为卷曲/链构象,从螺旋构象转变为卷曲/链构象。Leu、Ser、Thr 和 Val 更倾向于从链构象转变为卷曲/螺旋构象。
本研究获得的结果将有助于理解和预测蛋白质-蛋白质复合物中的结合位点。