Division of Biostatistics and Department of Statistics, University of California, Berkeley, USA.
BMC Bioinformatics. 2011 Dec 17;12:480. doi: 10.1186/1471-2105-12-480.
Transcriptome sequencing (RNA-Seq) has become the assay of choice for high-throughput studies of gene expression. However, as is the case with microarrays, major technology-related artifacts and biases affect the resulting expression measures. Normalization is therefore essential to ensure accurate inference of expression levels and subsequent analyses thereof.
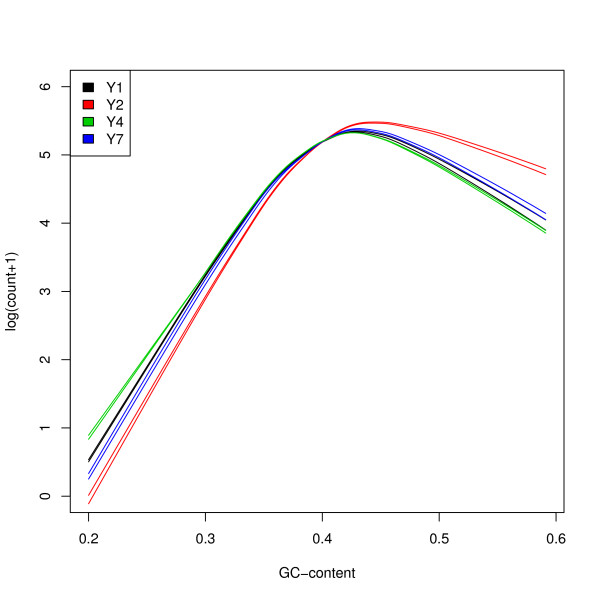
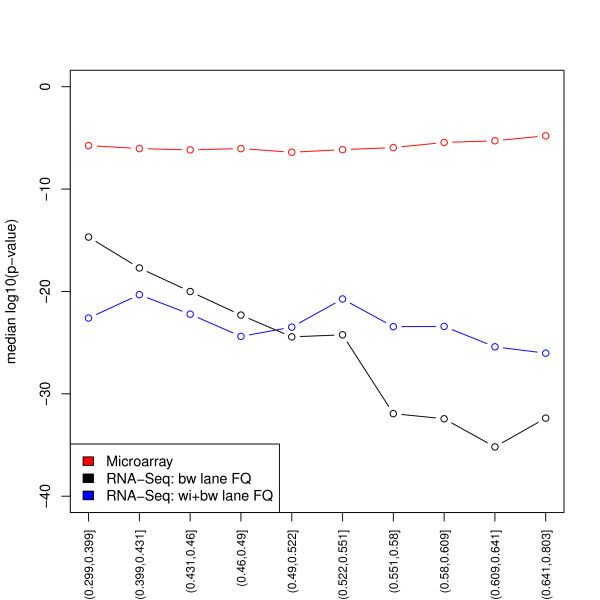
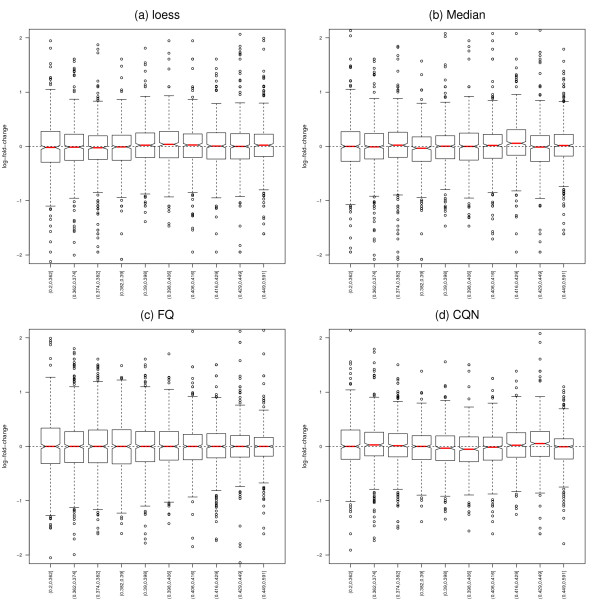
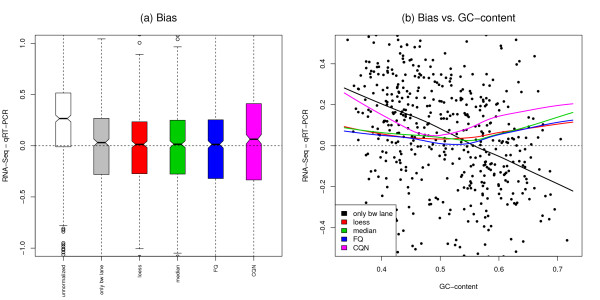
We focus on biases related to GC-content and demonstrate the existence of strong sample-specific GC-content effects on RNA-Seq read counts, which can substantially bias differential expression analysis. We propose three simple within-lane gene-level GC-content normalization approaches and assess their performance on two different RNA-Seq datasets, involving different species and experimental designs. Our methods are compared to state-of-the-art normalization procedures in terms of bias and mean squared error for expression fold-change estimation and in terms of Type I error and p-value distributions for tests of differential expression. The exploratory data analysis and normalization methods proposed in this article are implemented in the open-source Bioconductor R package EDASeq.
Our within-lane normalization procedures, followed by between-lane normalization, reduce GC-content bias and lead to more accurate estimates of expression fold-changes and tests of differential expression. Such results are crucial for the biological interpretation of RNA-Seq experiments, where downstream analyses can be sensitive to the supplied lists of genes.
转录组测序(RNA-Seq)已成为高通量基因表达研究的首选检测方法。然而,与微阵列一样,主要的技术相关伪影和偏差会影响到最终的表达测量结果。因此,为了确保对表达水平进行准确推断以及对后续分析,必须进行标准化。
我们专注于与 GC 含量相关的偏差,并证明在 RNA-Seq 读段计数上存在强烈的样本特异性 GC 含量效应,这可能会极大地影响差异表达分析。我们提出了三种简单的基于基因的 lane 内 GC 含量标准化方法,并在两个涉及不同物种和实验设计的不同 RNA-Seq 数据集上评估了它们的性能。我们的方法在表达倍数变化估计的偏差和均方误差方面,以及在差异表达检验的 Type I 错误和 p 值分布方面,与最先进的标准化程序进行了比较。本文中提出的探索性数据分析和标准化方法在开源 Bioconductor R 包 EDASeq 中实现。
我们的 lane 内标准化程序,再加上 lane 间标准化,减少了 GC 含量偏差,从而更准确地估计了表达倍数变化和差异表达检验。这些结果对于 RNA-Seq 实验的生物学解释至关重要,因为下游分析可能对提供的基因列表敏感。