Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA.
Biostatistics. 2012 Apr;13(2):204-16. doi: 10.1093/biostatistics/kxr054. Epub 2012 Jan 27.
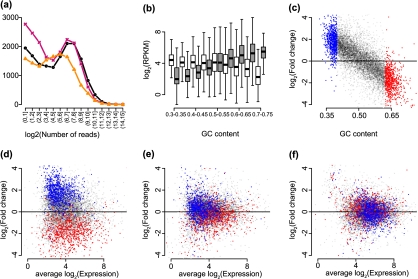
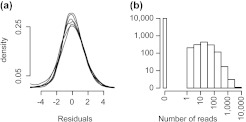
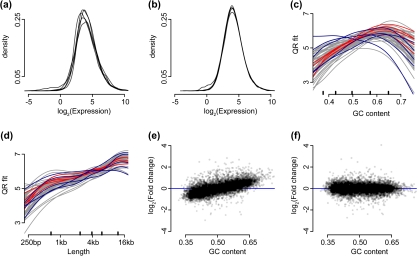
The ability to measure gene expression on a genome-wide scale is one of the most promising accomplishments in molecular biology. Microarrays, the technology that first permitted this, were riddled with problems due to unwanted sources of variability. Many of these problems are now mitigated, after a decade's worth of statistical methodology development. The recently developed RNA sequencing (RNA-seq) technology has generated much excitement in part due to claims of reduced variability in comparison to microarrays. However, we show that RNA-seq data demonstrate unwanted and obscuring variability similar to what was first observed in microarrays. In particular, we find guanine-cytosine content (GC-content) has a strong sample-specific effect on gene expression measurements that, if left uncorrected, leads to false positives in downstream results. We also report on commonly observed data distortions that demonstrate the need for data normalization. Here, we describe a statistical methodology that improves precision by 42% without loss of accuracy. Our resulting conditional quantile normalization algorithm combines robust generalized regression to remove systematic bias introduced by deterministic features such as GC-content and quantile normalization to correct for global distortions.
在全基因组范围内测量基因表达的能力是分子生物学最有前途的成就之一。微阵列技术是最初实现这一目标的技术,但由于存在不必要的变异源,存在许多问题。经过十年的统计方法学发展,许多这些问题现在得到了缓解。最近开发的 RNA 测序 (RNA-seq) 技术在部分由于与微阵列相比声称减少了变异性而引起了极大的关注。然而,我们表明,RNA-seq 数据显示出与最初在微阵列中观察到的类似的不需要的和掩盖的可变性。具体而言,我们发现鸟嘌呤-胞嘧啶含量 (GC 含量) 对基因表达测量具有强烈的样本特异性影响,如果不进行校正,会导致下游结果中的假阳性。我们还报告了常见的观察到的数据扭曲,这些扭曲表明需要数据归一化。在这里,我们描述了一种统计方法学,该方法在不损失准确性的情况下将精度提高了 42%。我们的条件分位数归一化算法结合了稳健的广义回归来消除 GC 含量等确定性特征引入的系统偏差,以及分位数归一化来纠正全局扭曲。