Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SA, UK.
Brief Bioinform. 2013 Mar;14(2):203-12. doi: 10.1093/bib/bbr073. Epub 2012 Jan 16.
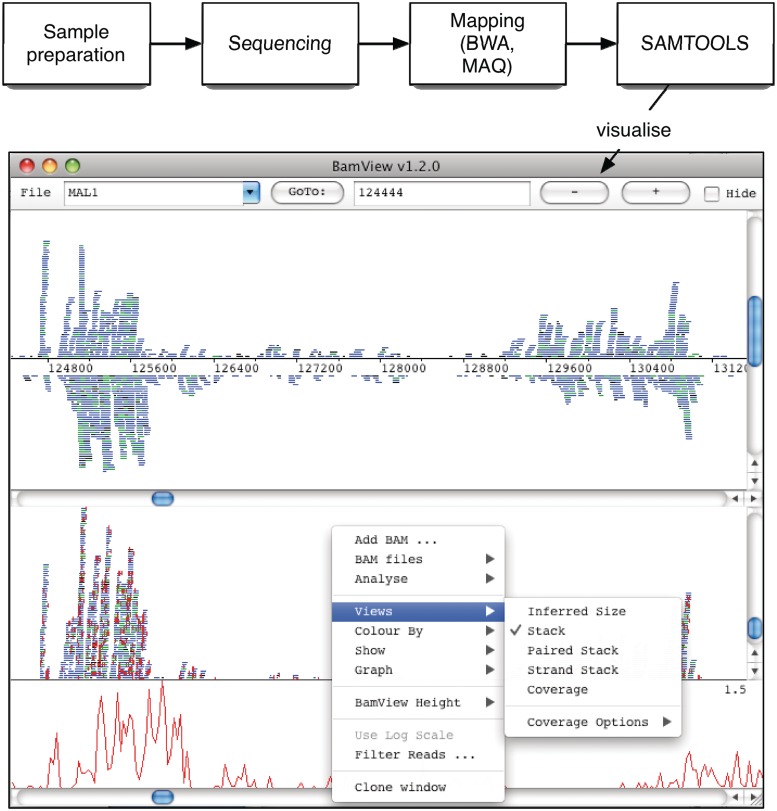
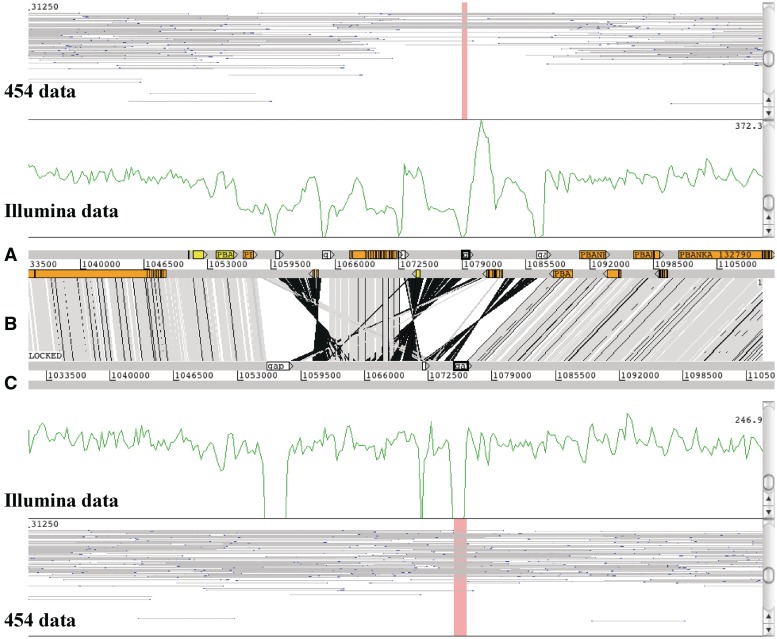
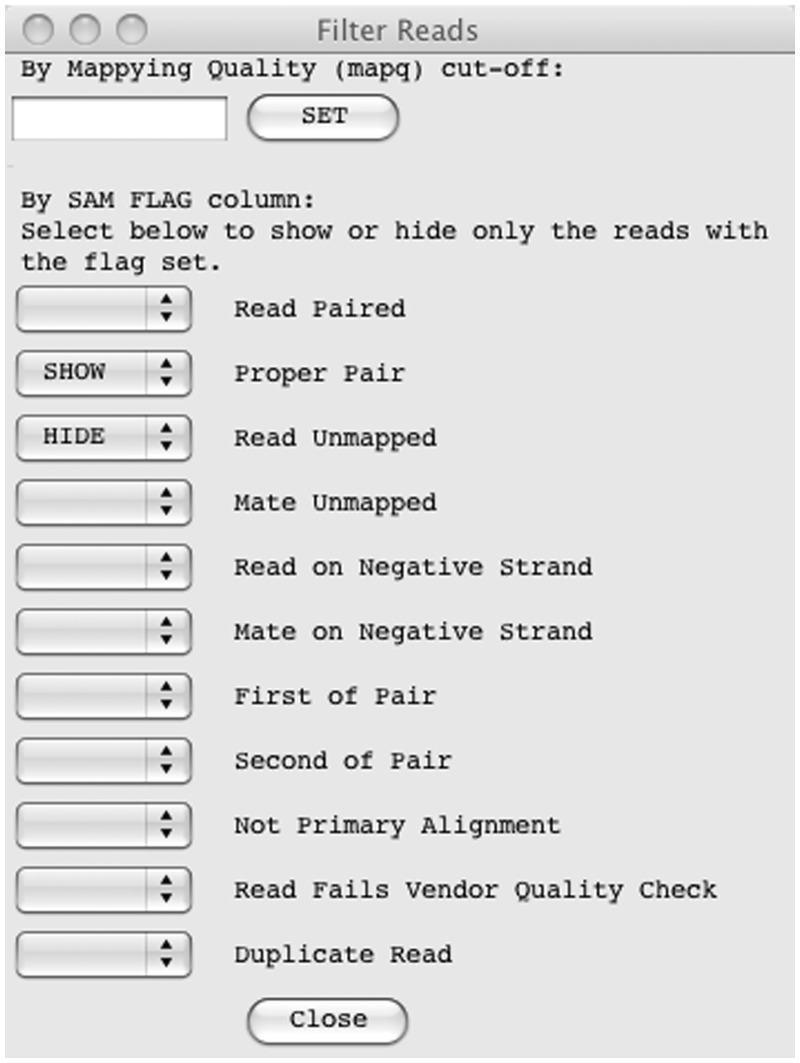
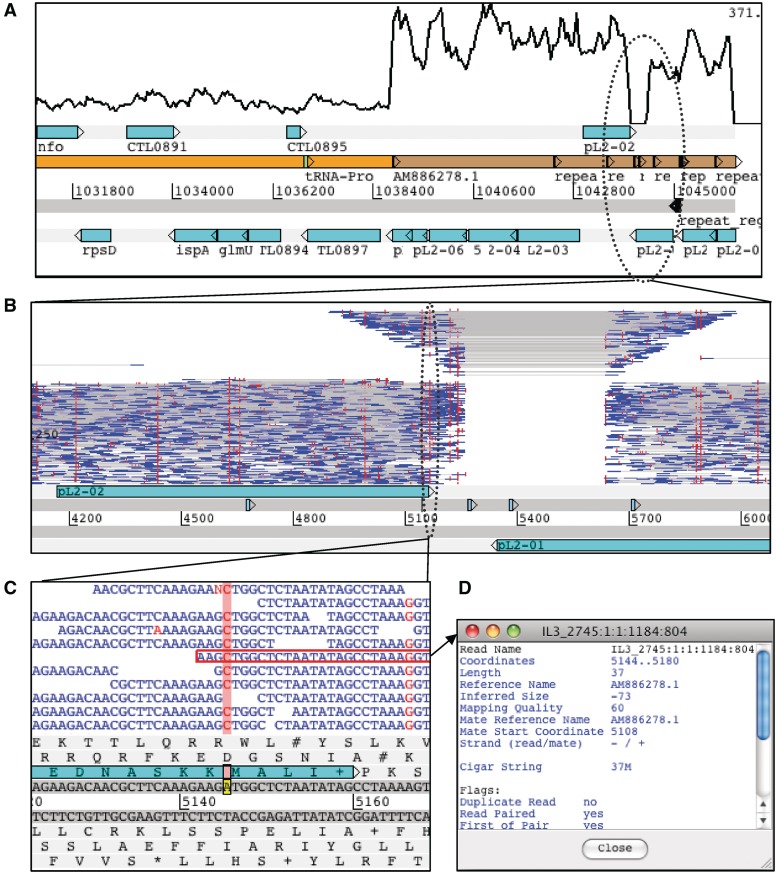
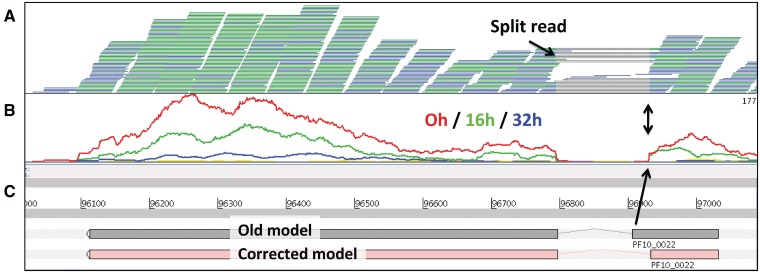
So-called next-generation sequencing (NGS) has provided the ability to sequence on a massive scale at low cost, enabling biologists to perform powerful experiments and gain insight into biological processes. BamView has been developed to visualize and analyse sequence reads from NGS platforms, which have been aligned to a reference sequence. It is a desktop application for browsing the aligned or mapped reads [Ruffalo, M, LaFramboise, T, Koyutürk, M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics 2011;27:2790-6] at different levels of magnification, from nucleotide level, where the base qualities can be seen, to genome or chromosome level where overall coverage is shown. To enable in-depth investigation of NGS data, various views are provided that can be configured to highlight interesting aspects of the data. Multiple read alignment files can be overlaid to compare results from different experiments, and filters can be applied to facilitate the interpretation of the aligned reads. As well as being a standalone application it can be used as an integrated part of the Artemis genome browser, BamView allows the user to study NGS data in the context of the sequence and annotation of the reference genome. Single nucleotide polymorphism (SNP) density and candidate SNP sites can be highlighted and investigated, and read-pair information can be used to discover large structural insertions and deletions. The application will also calculate simple analyses of the read mapping, including reporting the read counts and reads per kilobase per million mapped reads (RPKM) for genes selected by the user.
BamView and Artemis are freely available software. These can be downloaded from their home pages: http://bamview.sourceforge.net/; http://www.sanger.ac.uk/resources/software/artemis/. Requirements: Java 1.6 or higher.
所谓的下一代测序(NGS)已经提供了以低成本大规模测序的能力,使生物学家能够进行强大的实验,并深入了解生物过程。BamView 是为了可视化和分析来自 NGS 平台的序列读取而开发的,这些读取已经与参考序列对齐。它是一个用于在不同放大倍数下浏览对齐或映射读取的桌面应用程序 [Ruffalo,M,LaFramboise,T,Koyutürk,M. 下一代测序读取对齐算法的比较分析。生物信息学 2011;27:2790-6],从核苷酸水平,其中可以看到碱基质量,到基因组或染色体水平,显示整体覆盖范围。为了深入研究 NGS 数据,提供了各种视图,可以配置这些视图以突出显示数据的有趣方面。可以覆盖多个读取对齐文件,以比较来自不同实验的结果,并应用过滤器来帮助解释对齐的读取。BamView 不仅是一个独立的应用程序,还可以作为 Artemis 基因组浏览器的集成部分使用,允许用户在参考基因组的序列和注释的背景下研究 NGS 数据。可以突出显示和研究单核苷酸多态性(SNP)密度和候选 SNP 位点,并使用读取对信息来发现大的结构插入和缺失。该应用程序还将计算读取映射的简单分析,包括报告用户选择的基因的读取计数和每百万映射读取的每千碱基读取数(RPKM)。
BamView 和 Artemis 是免费提供的软件。可以从它们的主页下载:http://bamview.sourceforge.net/;http://www.sanger.ac.uk/resources/software/artemis/。要求:Java 1.6 或更高版本。