Center for Biomedical Informatics, The University of Chicago, Chicago, Illinois, United States of America.
PLoS Comput Biol. 2012 Jan;8(1):e1002350. doi: 10.1371/journal.pcbi.1002350. Epub 2012 Jan 26.
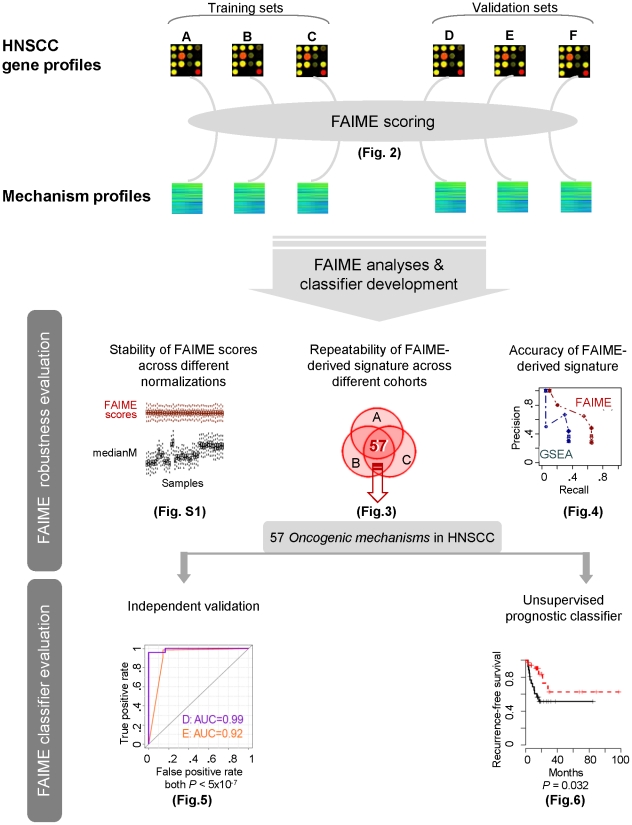
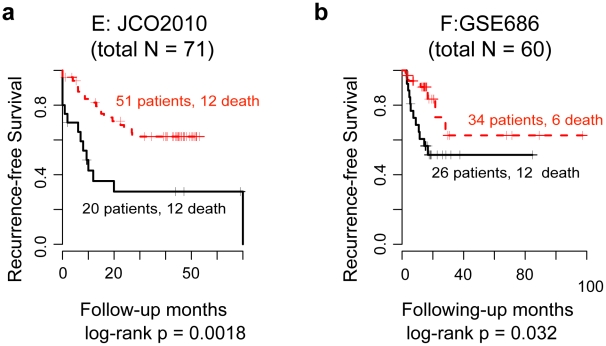
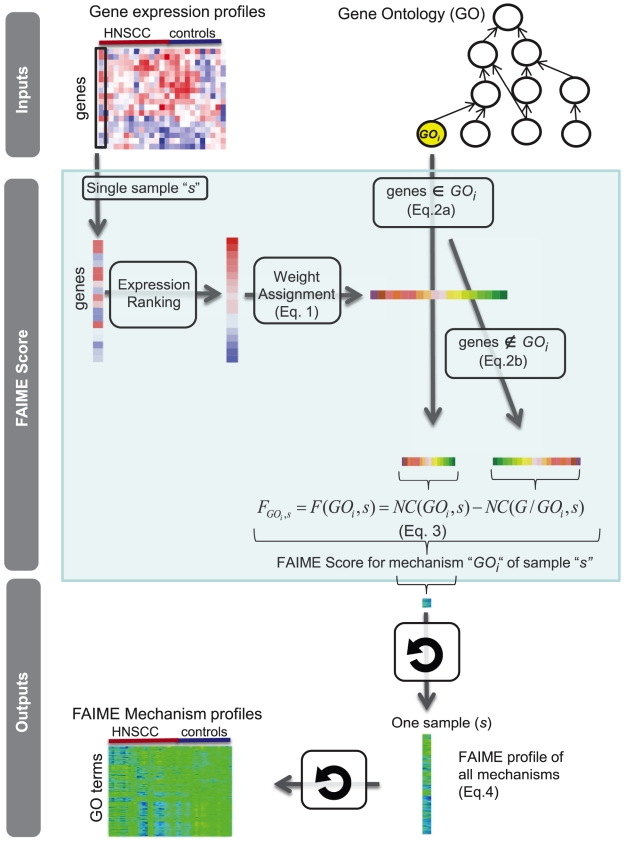
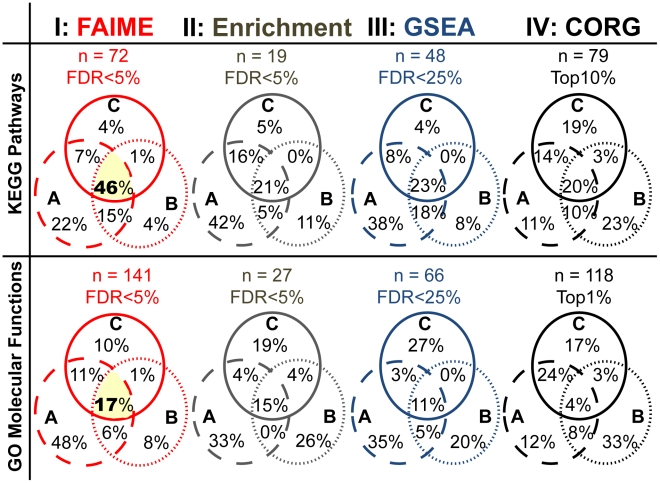
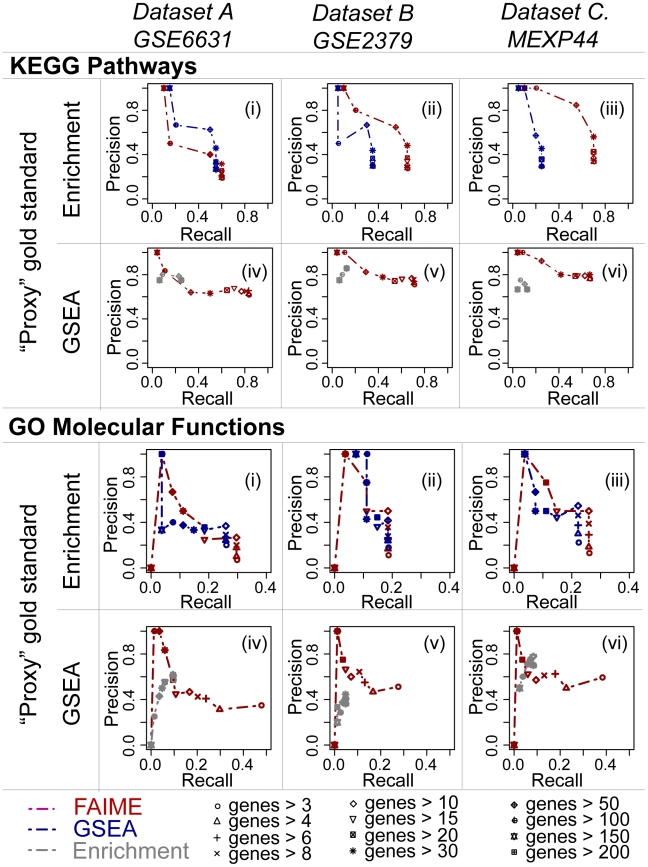
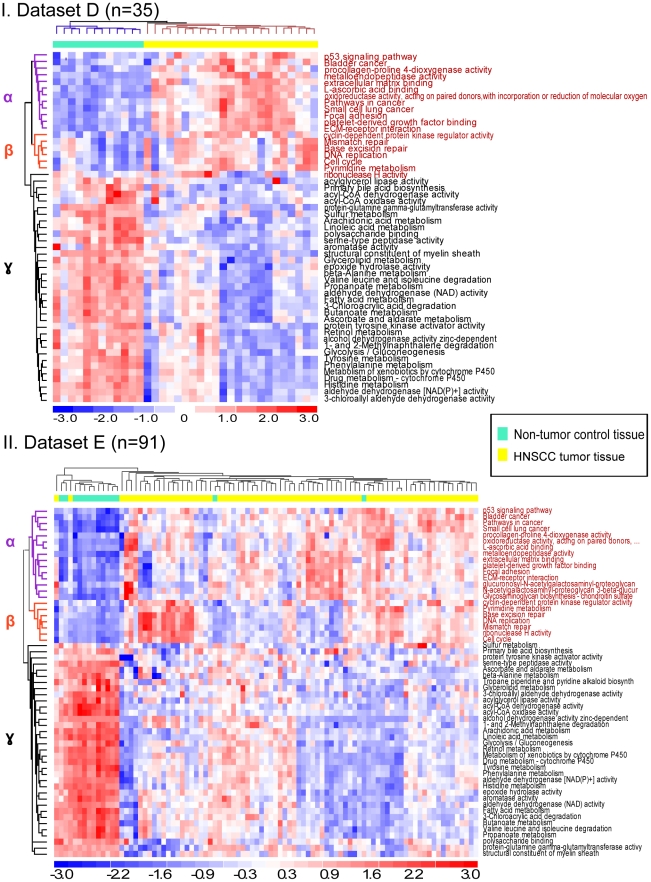
Gene expression signatures that are predictive of therapeutic response or prognosis are increasingly useful in clinical care; however, mechanistic (and intuitive) interpretation of expression arrays remains an unmet challenge. Additionally, there is surprisingly little gene overlap among distinct clinically validated expression signatures. These "causality challenges" hinder the adoption of signatures as compared to functionally well-characterized single gene biomarkers. To increase the utility of multi-gene signatures in survival studies, we developed a novel approach to generate "personal mechanism signatures" of molecular pathways and functions from gene expression arrays. FAIME, the Functional Analysis of Individual Microarray Expression, computes mechanism scores using rank-weighted gene expression of an individual sample. By comparing head and neck squamous cell carcinoma (HNSCC) samples with non-tumor control tissues, the precision and recall of deregulated FAIME-derived mechanisms of pathways and molecular functions are comparable to those produced by conventional cohort-wide methods (e.g. GSEA). The overlap of "Oncogenic FAIME Features of HNSCC" (statistically significant and differentially regulated FAIME-derived genesets representing GO functions or KEGG pathways derived from HNSCC tissue) among three distinct HNSCC datasets (pathways:46%, p<0.001) is more significant than the gene overlap (genes:4%). These Oncogenic FAIME Features of HNSCC can accurately discriminate tumors from control tissues in two additional HNSCC datasets (n = 35 and 91, F-accuracy = 100% and 97%, empirical p<0.001, area under the receiver operating characteristic curves = 99% and 92%), and stratify recurrence-free survival in patients from two independent studies (p = 0.0018 and p = 0.032, log-rank). Previous approaches depending on group assignment of individual samples before selecting features or learning a classifier are limited by design to discrete-class prediction. In contrast, FAIME calculates mechanism profiles for individual patients without requiring group assignment in validation sets. FAIME is more amenable for clinical deployment since it translates the gene-level measurements of each given sample into pathways and molecular function profiles that can be applied to analyze continuous phenotypes in clinical outcome studies (e.g. survival time, tumor volume).
基因表达谱可预测治疗反应或预后,在临床护理中越来越有用;然而,表达谱的机制(和直观)解释仍然是一个未满足的挑战。此外,不同临床验证的表达谱之间的基因重叠非常少。这些“因果关系挑战”阻碍了表达谱的采用,而功能良好的单个基因生物标志物则得到了广泛应用。为了提高多基因标志物在生存研究中的实用性,我们开发了一种从基因表达谱中生成分子途径和功能的“个体机制标志物”的新方法。FAIME(个体微阵列表达的功能分析)使用个体样本的秩加权基因表达计算机制评分。通过比较头颈部鳞状细胞癌(HNSCC)样本和非肿瘤对照组织,FAIME 衍生的途径和分子功能失调机制的精确性和召回率与传统的全队列方法(例如 GSEA)相当。三个不同的 HNSCC 数据集之间的“HNSCC 的致癌 FAIME 特征”(代表 HNSCC 组织衍生的 GO 功能或 KEGG 途径的统计学显著和差异调节的 FAIME 衍生基因集)的重叠(通路:46%,p<0.001)比基因重叠(基因:4%)更显著。这些 HNSCC 的致癌 FAIME 特征可以在另外两个 HNSCC 数据集(n = 35 和 91,F-准确性=100%和 97%,经验 p<0.001,接受者操作特征曲线下面积=99%和 92%)中准确地区分肿瘤和对照组织,并分层两个独立研究中的无复发生存(p=0.0018 和 p=0.032,对数秩)。以前的方法在选择特征或学习分类器之前依赖于个体样本的分组,因此在设计上仅限于离散类别的预测。相比之下,FAIME 无需在验证集中进行分组即可为个体患者计算机制谱。FAIME 更适合临床部署,因为它将每个给定样本的基因水平测量值转换为可以应用于分析临床结果研究(例如生存时间、肿瘤体积)中连续表型的途径和分子功能谱。