Department of Human Genetics, McGill University, Montreal, Canada.
PLoS Genet. 2012 Feb;8(2):e1002496. doi: 10.1371/journal.pgen.1002496. Epub 2012 Feb 2.
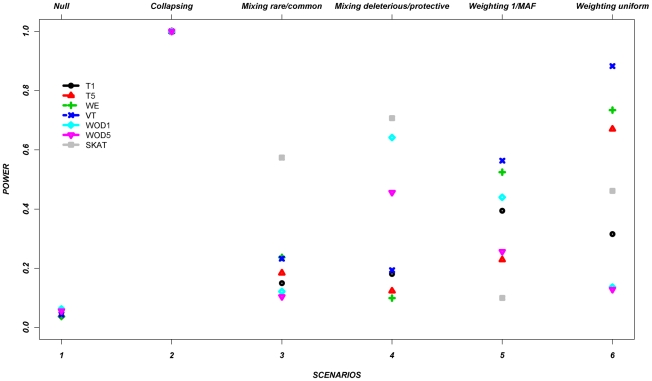
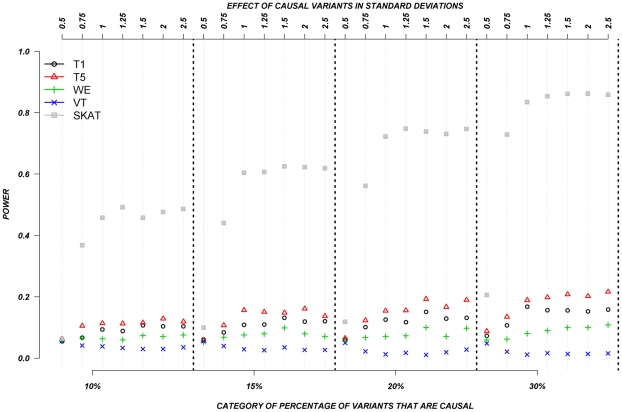
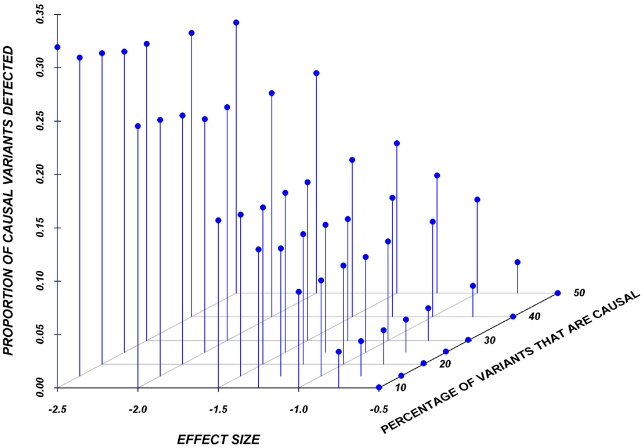
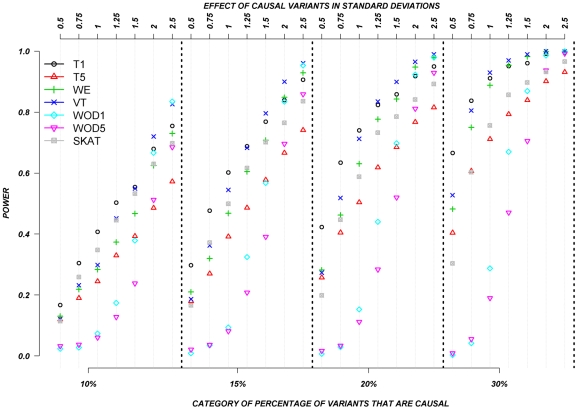
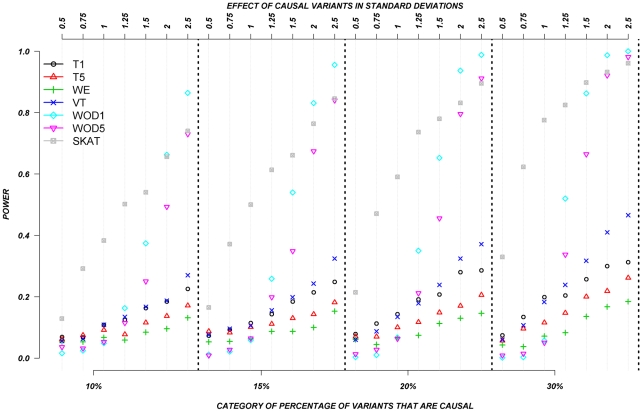
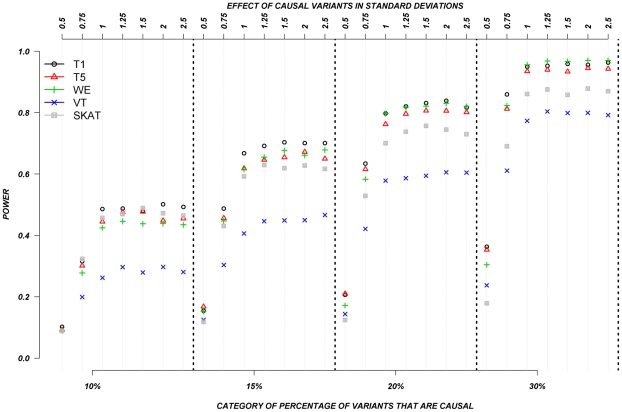
The role of rare genetic variation in the etiology of complex disease remains unclear. However, the development of next-generation sequencing technologies offers the experimental opportunity to address this question. Several novel statistical methodologies have been recently proposed to assess the contribution of rare variation to complex disease etiology. Nevertheless, no empirical estimates comparing their relative power are available. We therefore assessed the parameters that influence their statistical power in 1,998 individuals Sanger-sequenced at seven genes by modeling different distributions of effect, proportions of causal variants, and direction of the associations (deleterious, protective, or both) in simulated continuous trait and case/control phenotypes. Our results demonstrate that the power of recently proposed statistical methods depend strongly on the underlying hypotheses concerning the relationship of phenotypes with each of these three factors. No method demonstrates consistently acceptable power despite this large sample size, and the performance of each method depends upon the underlying assumption of the relationship between rare variants and complex traits. Sensitivity analyses are therefore recommended to compare the stability of the results arising from different methods, and promising results should be replicated using the same method in an independent sample. These findings provide guidance in the analysis and interpretation of the role of rare base-pair variation in the etiology of complex traits and diseases.
稀有遗传变异在复杂疾病病因学中的作用仍不清楚。然而,下一代测序技术的发展为解决这个问题提供了实验机会。最近提出了几种新的统计方法来评估稀有变异对复杂疾病病因的贡献。然而,尚无比较它们相对功效的经验估计。因此,我们通过模拟不同的效应分布、因果变异的比例以及连续性状和病例/对照表型中关联的方向(有害、保护或两者兼有),在 7 个基因的 1998 个桑格测序个体中评估了影响其统计功效的参数。我们的结果表明,最近提出的统计方法的功效强烈依赖于关于表型与这三个因素中每一个之间关系的基本假设。尽管样本量很大,但没有一种方法表现出一致可接受的功效,而且每种方法的性能都取决于稀有变异与复杂性状之间关系的基本假设。因此,建议进行敏感性分析以比较不同方法产生的结果的稳定性,并且应该使用相同的方法在独立样本中复制有希望的结果。这些发现为分析和解释稀有碱基对变异在复杂性状和疾病病因学中的作用提供了指导。