Bioinformatics and Systems Engineering division, RIKEN Yokohama Institute, Tsurumi, Yokohama, Kanagawa 230-0045, Japan.
Bioinformatics. 2012 Apr 1;28(7):929-37. doi: 10.1093/bioinformatics/bts065. Epub 2012 Feb 13.
A reconstruction of full-length transcripts observed by next-generation sequencer or tiling arrays is an essential technique to know all phenomena of transcriptomes. Several techniques of the reconstruction have been developed. However, problems of high-level noises and biases still remain and interrupt the reconstruction. A method is required that is robust against noise and bias and correctly reconstructs transcripts regardless of equipment used.
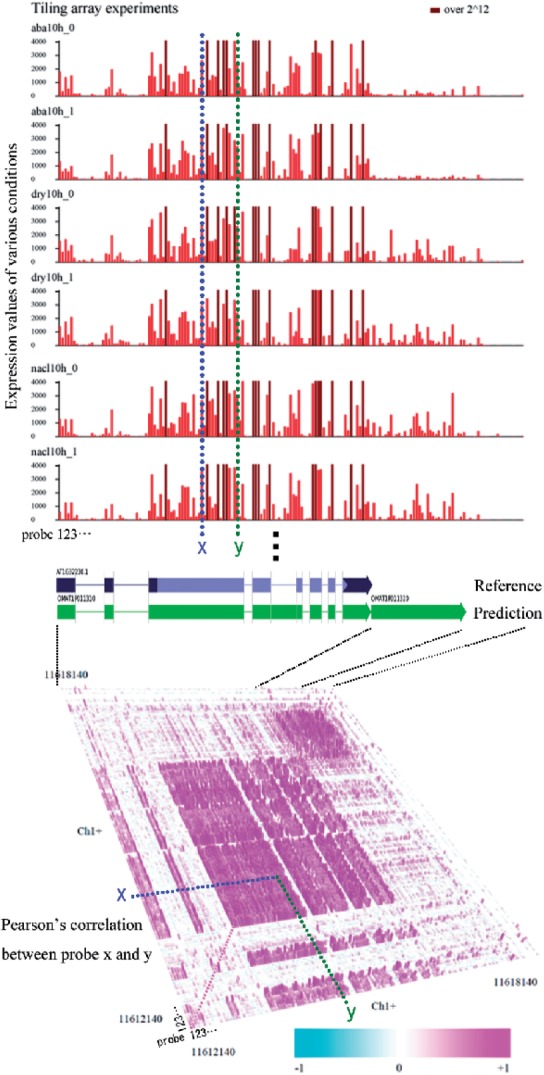
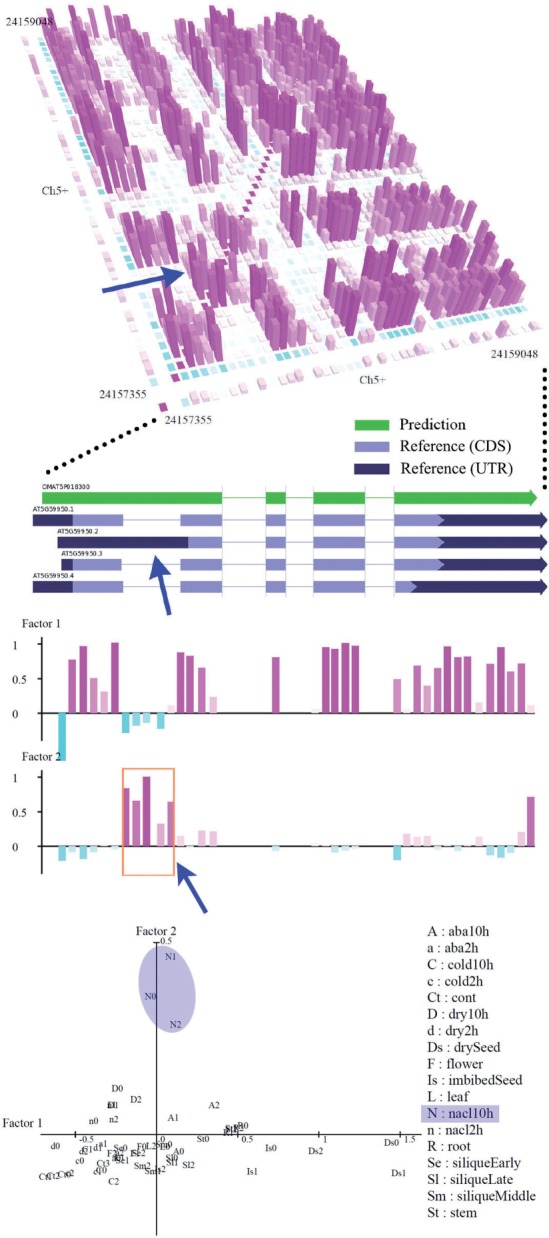
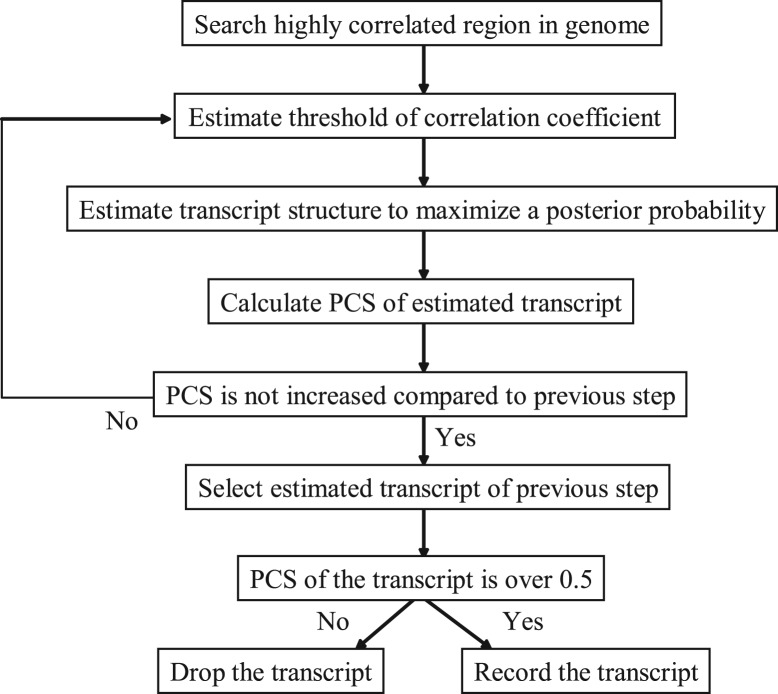
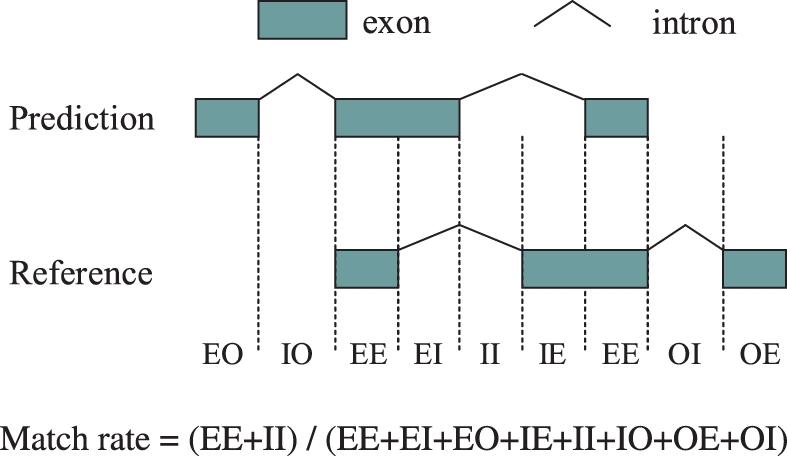
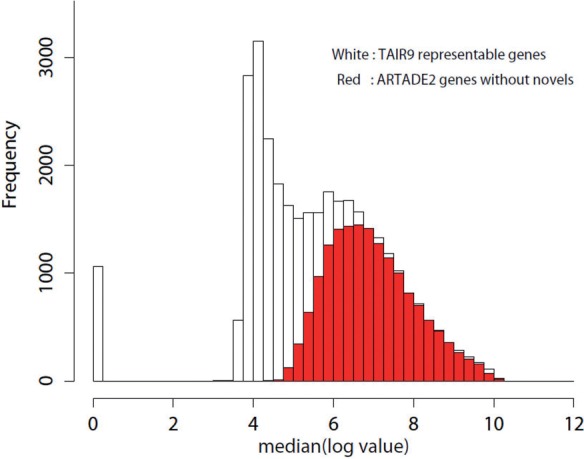
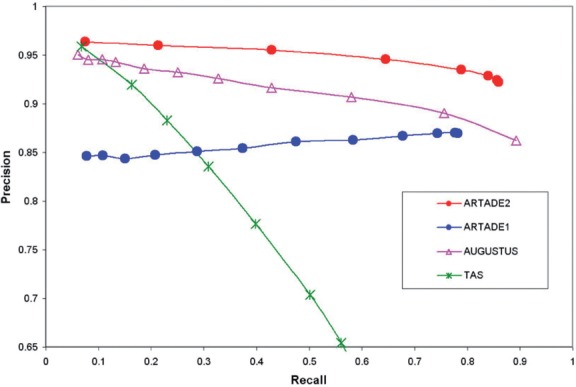
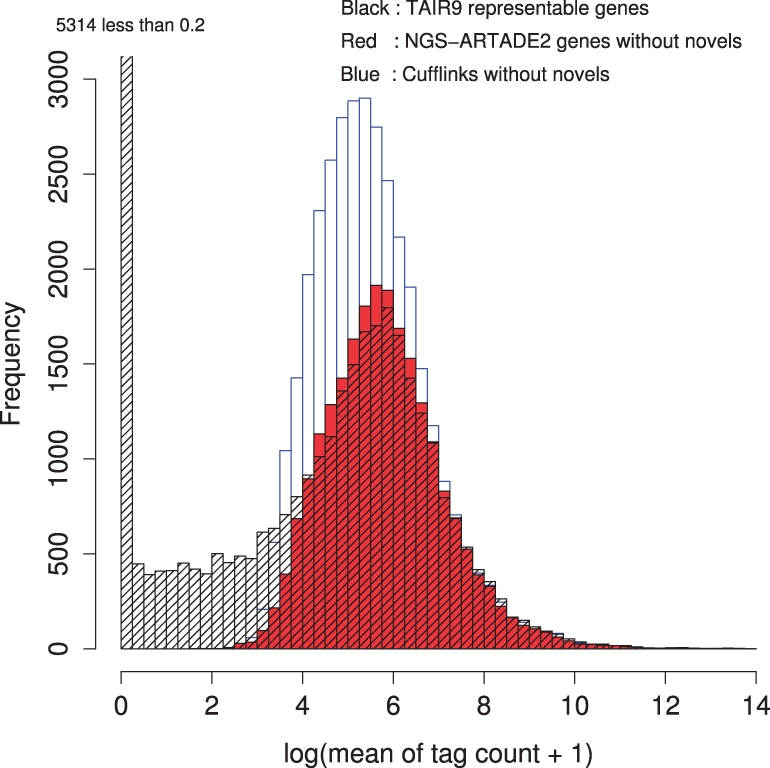
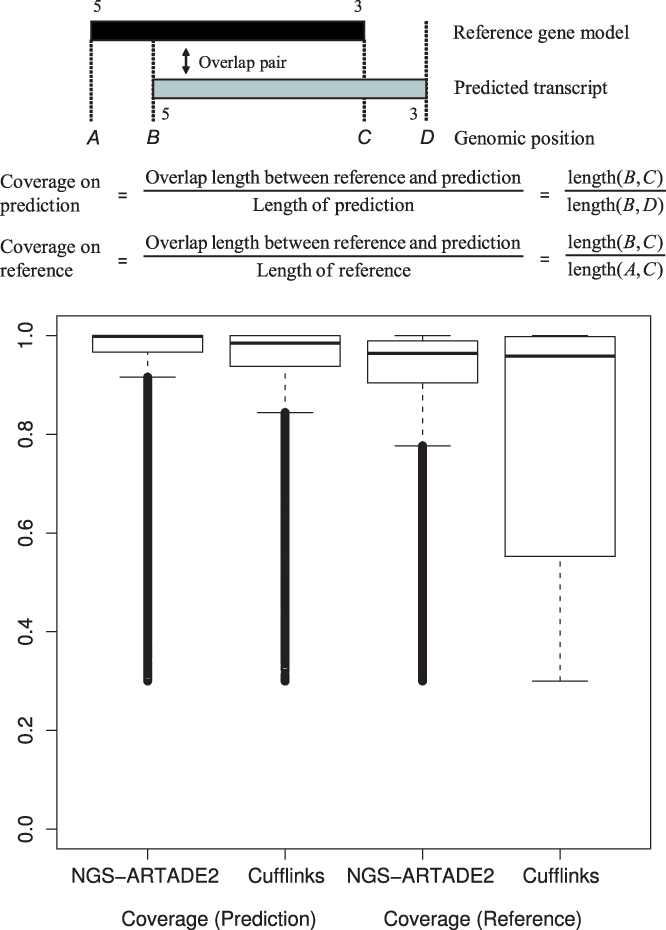
We propose a completely new statistical method that reconstructs full-length transcripts and can be applied on both next-generation sequencers and tiling arrays. The method called ARTADE2 analyzes 'positional correlation', meaning correlations of expression values for every combination on genomic positions of multiple transcriptional data. ARTADE2 then reconstructs full-length transcripts using a logistic model based on the positional correlation and the Markov model. ARTADE2 elucidated 17 591 full-length transcripts from 55 transcriptome datasets and showed notable performance compared with other recent prediction methods. Moreover, 1489 novel transcripts were discovered. We experimentally tested 16 novel transcripts, among which 14 were confirmed by reverse transcription-polymerase chain reaction and sequence mapping. The method also showed notable performance for reconstructing of mRNA observed by a next-generation sequencer. Moreover, the positional correlation and factor analysis embedded in ARTADE2 successfully detected regions at which alternative isoforms may exist, and thus are expected to be applied for discovering transcript biomarkers for a wide range of disciplines including preemptive medicine.
Supplementary data are available at Bioinformatics online.
通过下一代测序仪或平铺阵列观察到全长转录本的重建是了解转录组所有现象的重要技术。已经开发了几种重建技术。然而,高水平噪声和偏差的问题仍然存在,并中断了重建。需要一种能够抵抗噪声和偏差并正确重建转录本的方法,而与所使用的设备无关。
我们提出了一种全新的统计方法,该方法可以重建全长转录本,并可应用于下一代测序仪和平铺阵列。该方法称为 ARTADE2,分析了“位置相关性”,即多个转录组数据的基因组位置上的表达值的组合的相关性。然后,ARTADE2 使用基于位置相关性和马尔可夫模型的逻辑模型来重建全长转录本。ARTADE2 从 55 个转录组数据集重建了 17591 个全长转录本,并与其他最近的预测方法相比表现出了显著的性能。此外,还发现了 1489 个新的转录本。我们通过实验测试了 16 个新的转录本,其中 14 个通过逆转录-聚合酶链反应和序列映射得到了证实。该方法还显示出在重建下一代测序仪观察到的 mRNA 方面的显著性能。此外,ARTADE2 中嵌入的位置相关性和因子分析成功地检测到了可能存在替代异构体的区域,因此有望应用于发现包括预防性医学在内的广泛学科的转录生物标志物。
补充数据可在生物信息学在线获得。