Department of Biochemistry and Molecular Genetics, University of Colorado School of Medicine, Aurora, Colorado, United States of America.
PLoS One. 2012;7(2):e30953. doi: 10.1371/journal.pone.0030953. Epub 2012 Feb 14.
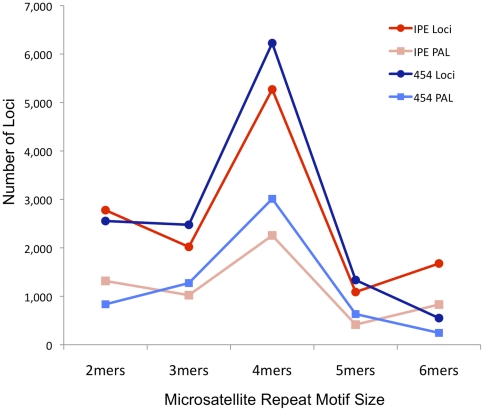
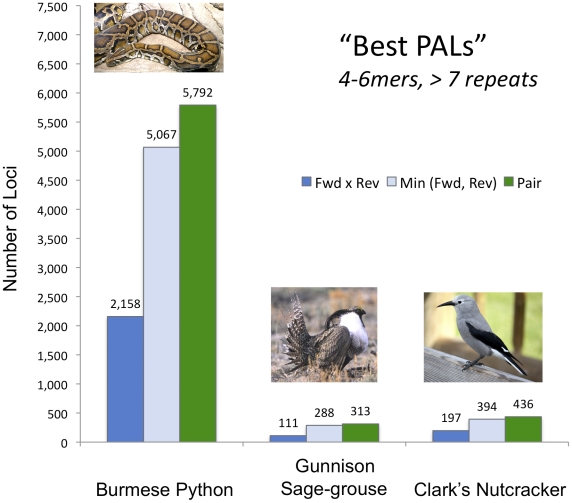
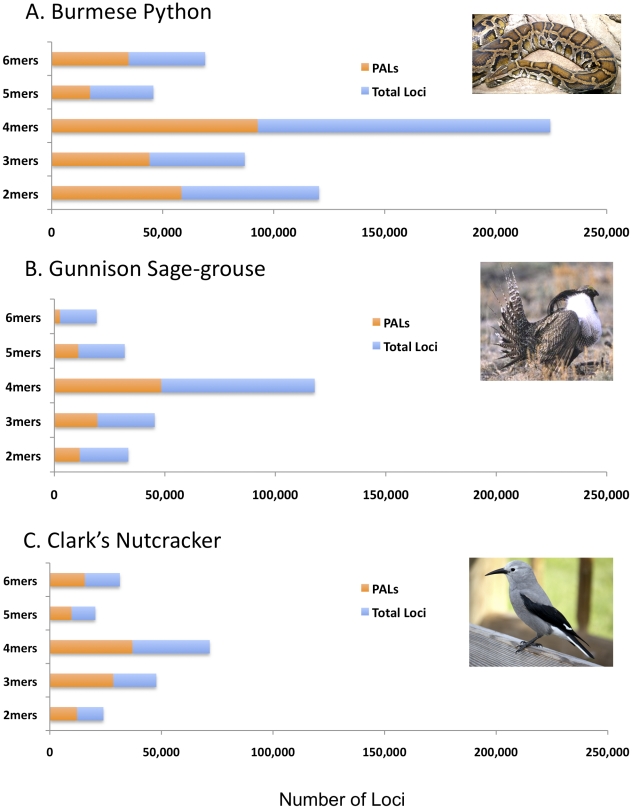
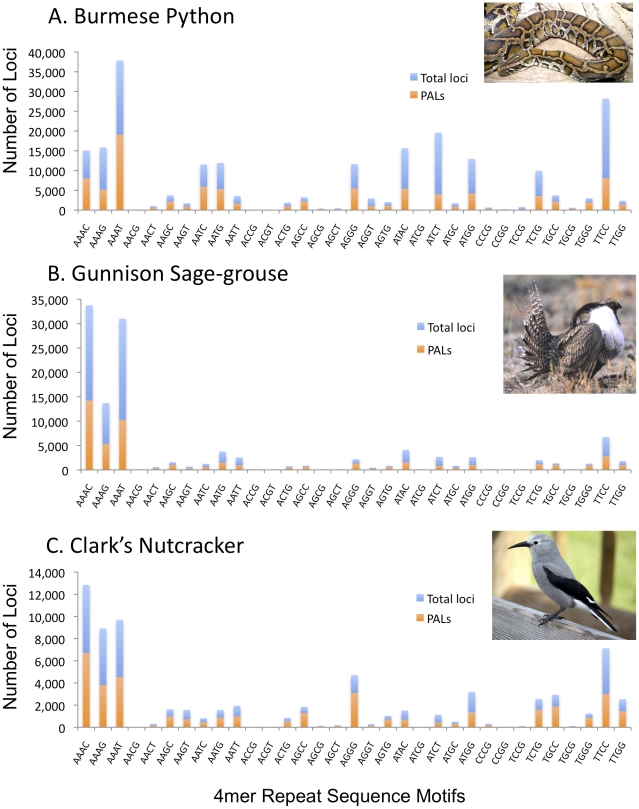
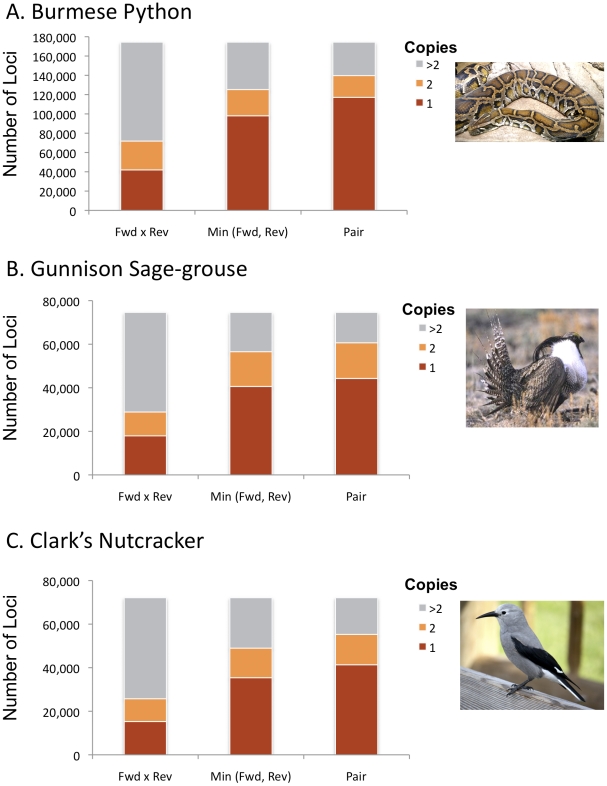
Identification of microsatellites, or simple sequence repeats (SSRs), can be a time-consuming and costly investment requiring enrichment, cloning, and sequencing of candidate loci. Recently, however, high throughput sequencing (with or without prior enrichment for specific SSR loci) has been utilized to identify SSR loci. The direct "Seq-to-SSR" approach has an advantage over enrichment-based strategies in that it does not require a priori selection of particular motifs, or prior knowledge of genomic SSR content. It has been more expensive per SSR locus recovered, however, particularly for genomes with few SSR loci, such as bird genomes. The longer but relatively more expensive 454 reads have been preferred over less expensive Illumina reads. Here, we use Illumina paired-end sequence data to identify potentially amplifiable SSR loci (PALs) from a snake (the Burmese python, Python molurus bivittatus), and directly compare these results to those from 454 data. We also compare the python results to results from Illumina sequencing of two bird genomes (Gunnison Sage-grouse, Centrocercus minimus, and Clark's Nutcracker, Nucifraga columbiana), which have considerably fewer SSRs than the python. We show that direct Illumina Seq-to-SSR can identify and characterize thousands of potentially amplifiable SSR loci for as little as $10 per sample--a fraction of the cost of 454 sequencing. Given that Illumina Seq-to-SSR is effective, inexpensive, and reliable even for species such as birds that have few SSR loci, it seems that there are now few situations for which prior hybridization is justifiable.
微卫星或简单重复序列 (SSR) 的鉴定可能是一项耗时且昂贵的投资,需要对候选基因座进行富集、克隆和测序。然而,最近高通量测序(无论是否事先对特定 SSR 基因座进行富集)已被用于鉴定 SSR 基因座。与基于富集的策略相比,直接“测序到 SSR”方法具有优势,因为它不需要对特定基序进行先验选择,也不需要事先了解基因组 SSR 含量。然而,与富集策略相比,它每鉴定一个 SSR 基因座的成本更高,特别是对于 SSR 基因座较少的基因组,如鸟类基因组。尽管 454 读长较长但相对昂贵,但它比更便宜的 Illumina 读长更受欢迎。在这里,我们使用 Illumina 配对末端测序数据从蛇(缅甸蟒,Python molurus bivittatus)中鉴定潜在可扩增 SSR 基因座(PAL),并直接将这些结果与 454 数据的结果进行比较。我们还将 python 的结果与两个鸟类基因组(Gunnison Sage-grouse,Centrocercus minimus 和 Clark's Nutcracker,Nucifraga columbiana)的 Illumina 测序结果进行比较,这两个鸟类基因组的 SSR 数量明显少于 python。我们表明,直接的 Illumina Seq-to-SSR 可以鉴定和表征数千个潜在可扩增的 SSR 基因座,每个样本的成本仅为 10 美元——这只是 454 测序成本的一小部分。鉴于 Illumina Seq-to-SSR 即使对于 SSR 基因座较少的鸟类等物种也非常有效、廉价且可靠,似乎现在很少有情况需要事先进行杂交。