Department of Plant Biology and Pathology, School of Environmental and Biological Sciences, Rutgers The State University of New Jersey, New Brunswick, New Jersey, United States of America.
PLoS One. 2013 Nov 27;8(11):e82408. doi: 10.1371/journal.pone.0082408. eCollection 2013.
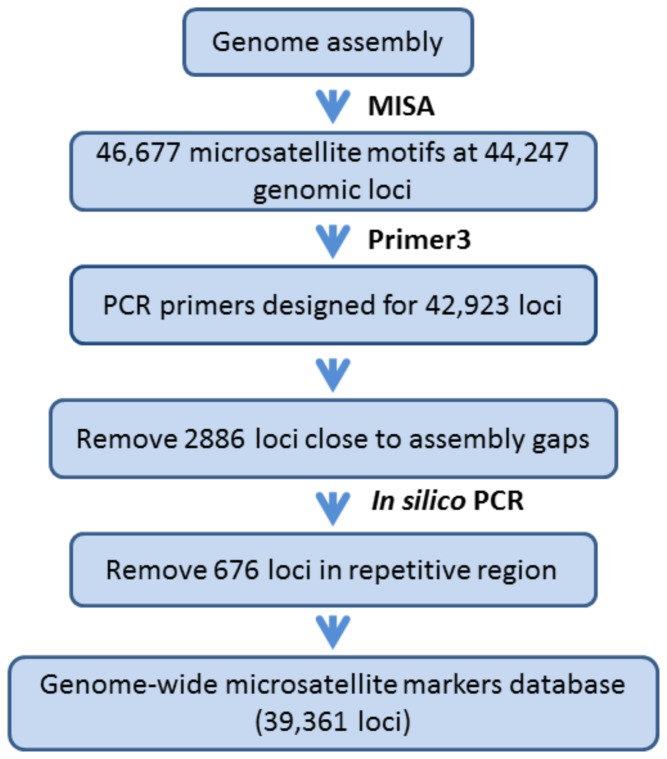
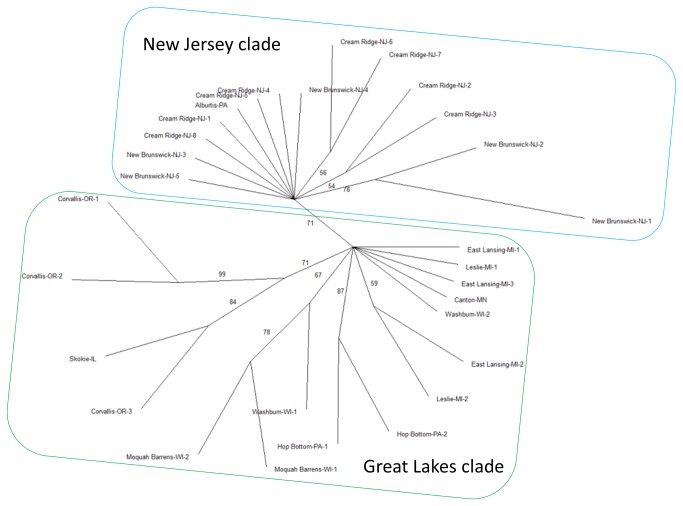
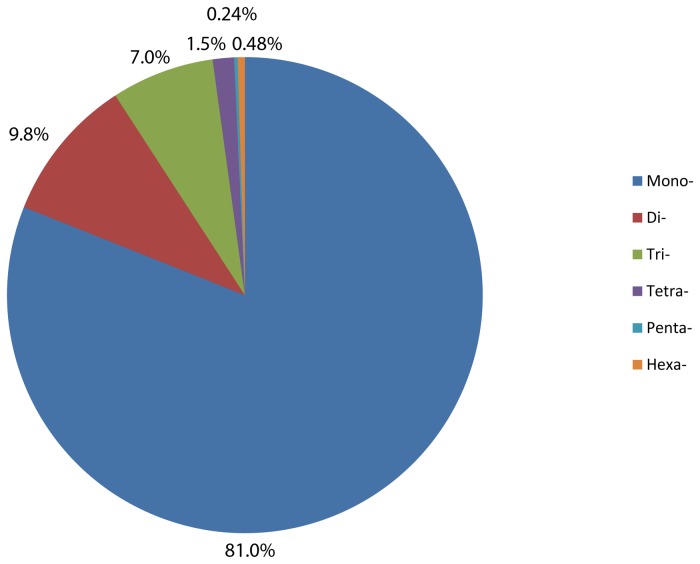
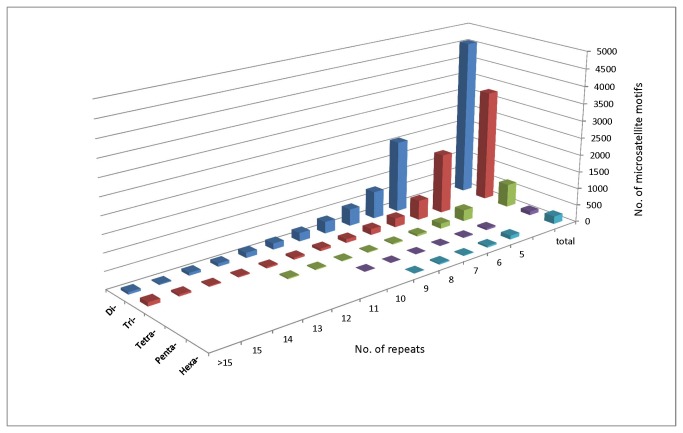
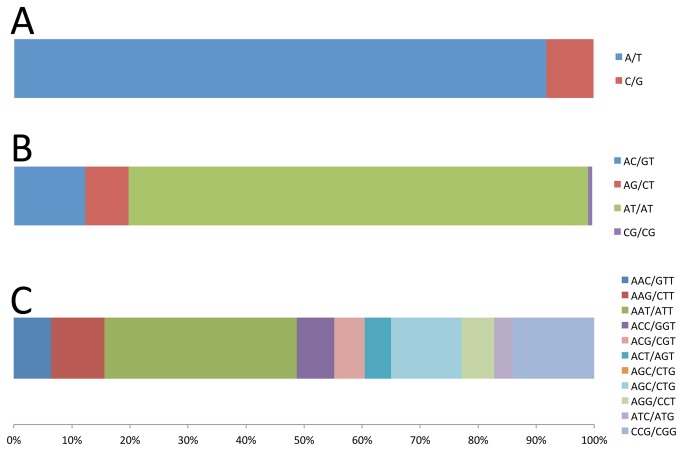
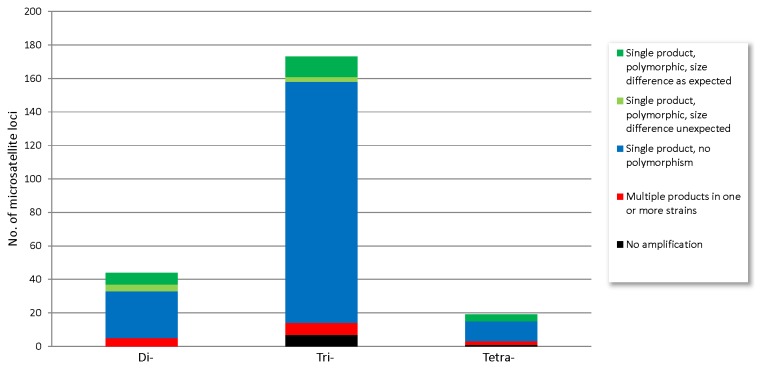
High-throughput sequencing has been dramatically accelerating the discovery of microsatellite markers (also known as Simple Sequence Repeats). Both 454 and Illumina reads have been used directly in microsatellite discovery and primer design (the "Seq-to-SSR" approach). However, constraints of this approach include: 1) many microsatellite-containing reads do not have sufficient flanking sequences to allow primer design, and 2) difficulties in removing microsatellite loci residing in longer, repetitive regions. In the current study, we applied the novel "Seq-Assembly-SSR" approach to overcome these constraints in Anisogramma anomala. In our approach, Illumina reads were first assembled into a draft genome, and the latter was then used in microsatellite discovery. A. anomala is an obligate biotrophic ascomycete that causes eastern filbert blight disease of commercial European hazelnut. Little is known about its population structure or diversity. Approximately 26 M 146 bp Illumina reads were generated from a paired-end library of a fungal strain from Oregon. The reads were assembled into a draft genome of 333 Mb (excluding gaps), with contig N50 of 10,384 bp and scaffold N50 of 32,987 bp. A bioinformatics pipeline identified 46,677 microsatellite motifs at 44,247 loci, including 2,430 compound loci. Primers were successfully designed for 42,923 loci (97%). After removing 2,886 loci close to assembly gaps and 676 loci in repetitive regions, a genome-wide microsatellite database of 39,361 loci was generated for the fungus. In experimental screening of 236 loci using four geographically representative strains, 228 (96.6%) were successfully amplified and 214 (90.7%) produced single PCR products. Twenty-three (9.7%) were found to be perfect polymorphic loci. A small-scale population study using 11 polymorphic loci revealed considerable gene diversity. Clustering analysis grouped isolates of this fungus into two clades in accordance with their geographic origins. Thus, the "Seq-Assembly-SSR" approach has proven to be a successful one for microsatellite discovery.
高通量测序极大地加速了微卫星标记(也称为简单序列重复)的发现。454 和 Illumina 读取序列都已直接用于微卫星发现和引物设计(“Seq-to-SSR”方法)。然而,这种方法的局限性包括:1)许多包含微卫星的读取序列没有足够的侧翼序列来允许引物设计,以及 2)在去除位于较长重复区域的微卫星位点方面存在困难。在本研究中,我们应用了新颖的“Seq-Assembly-SSR”方法来克服 Anisogramma anomala 中的这些限制。在我们的方法中,首先将 Illumina 读取序列组装成一个草图基因组,然后在微卫星发现中使用后者。A. anomala 是一种专性生物营养的子囊菌,会导致商业欧洲榛树的东部榛枯萎病。关于它的种群结构或多样性知之甚少。大约 26 M 146 bp Illumina 读取序列来自俄勒冈州真菌菌株的一个配对末端文库。这些读取序列被组装成一个 333 Mb 的草图基因组(不包括缺口),contig N50 为 10,384 bp,scaffold N50 为 32,987 bp。一个生物信息学管道在 44,247 个位点上识别出 46,677 个微卫星基序,包括 2,430 个复合位点。成功设计了 42,923 个位点(97%)的引物。在去除 2,886 个靠近组装缺口的位点和 676 个重复区域的位点后,生成了该真菌的 39,361 个位点的全基因组微卫星数据库。在使用四个具有地理代表性的菌株对 236 个位点进行的实验筛选中,成功扩增了 228 个位点(96.6%),214 个位点(90.7%)产生了单 PCR 产物。发现 23 个位点(9.7%)是完美的多态性位点。使用 11 个多态性位点的小规模种群研究显示出相当大的基因多样性。聚类分析根据地理来源将真菌的分离株分为两个分支。因此,“Seq-Assembly-SSR”方法已被证明是一种成功的微卫星发现方法。