Department of General Biology, Federal University of Minas Gerais, Belo Horizonte, Minas Gerais, Brazil.
PLoS One. 2012;7(2):e30848. doi: 10.1371/journal.pone.0030848. Epub 2012 Feb 15.
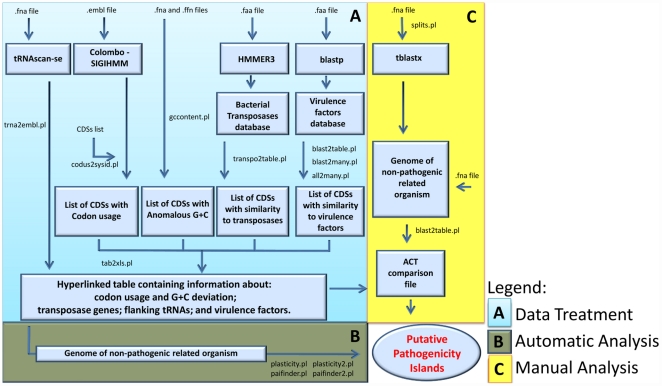
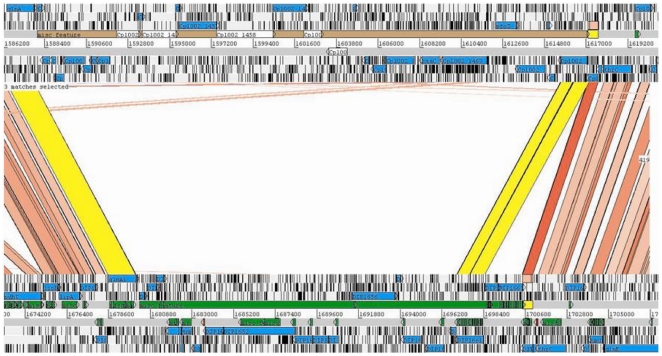
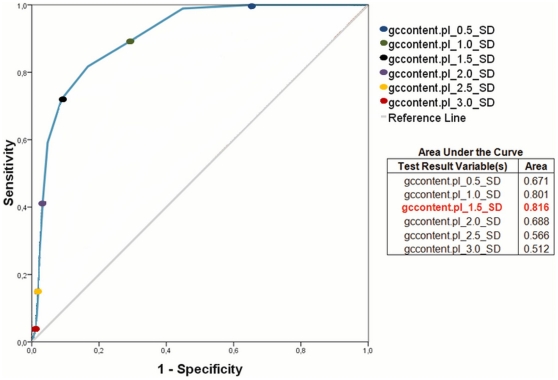
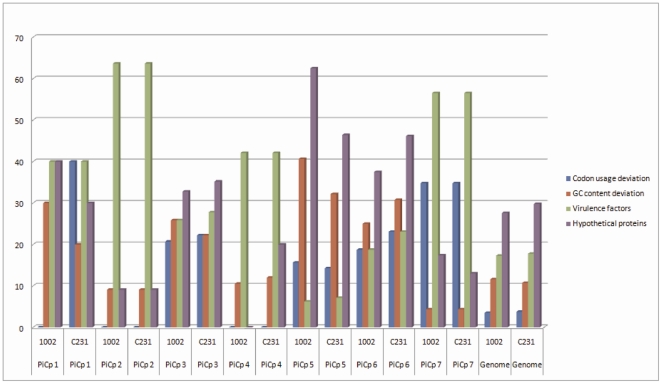
The adaptability of pathogenic bacteria to hosts is influenced by the genomic plasticity of the bacteria, which can be increased by such mechanisms as horizontal gene transfer. Pathogenicity islands play a major role in this type of gene transfer because they are large, horizontally acquired regions that harbor clusters of virulence genes that mediate the adhesion, colonization, invasion, immune system evasion, and toxigenic properties of the acceptor organism. Currently, pathogenicity islands are mainly identified in silico based on various characteristic features: (1) deviations in codon usage, G+C content or dinucleotide frequency and (2) insertion sequences and/or tRNA genetic flanking regions together with transposase coding genes. Several computational techniques for identifying pathogenicity islands exist. However, most of these techniques are only directed at the detection of horizontally transferred genes and/or the absence of certain genomic regions of the pathogenic bacterium in closely related non-pathogenic species. Here, we present a novel software suite designed for the prediction of pathogenicity islands (pathogenicity island prediction software, or PIPS). In contrast to other existing tools, our approach is capable of utilizing multiple features for pathogenicity island detection in an integrative manner. We show that PIPS provides better accuracy than other available software packages. As an example, we used PIPS to study the veterinary pathogen Corynebacterium pseudotuberculosis, in which we identified seven putative pathogenicity islands.
细菌对宿主的适应性受到其基因组可塑性的影响,这种可塑性可以通过水平基因转移等机制来增加。致病岛在这种基因转移中起着重要作用,因为它们是大型的、水平获得的区域,其中包含簇状毒力基因,这些基因介导了受体生物的粘附、定植、入侵、免疫系统逃避和产毒特性。目前,主要通过各种特征在计算机上识别致病岛:(1)密码子使用、G+C 含量或二核苷酸频率的偏差,以及(2)插入序列和/或 tRNA 遗传侧翼区域以及转座酶编码基因。存在几种用于识别致病岛的计算技术。然而,这些技术中的大多数仅针对水平转移基因的检测和/或在密切相关的非致病物种中缺乏致病性细菌的某些基因组区域。在这里,我们提出了一种用于预测致病岛的新软件套件(致病性岛预测软件,或 PIPS)。与其他现有工具不同,我们的方法能够以集成的方式利用多种特征来进行致病岛检测。我们表明 PIPS 提供了比其他可用软件包更高的准确性。例如,我们使用 PIPS 研究了兽医病原体棒状杆菌,在其中我们鉴定了七个假定的致病岛。