Division of Gastroenterology, Department of Internal Medicine, Baylor Research Institute, Baylor University Medical Center, Dallas, Texas, United States of America.
PLoS One. 2012;7(2):e31231. doi: 10.1371/journal.pone.0031231. Epub 2012 Feb 17.
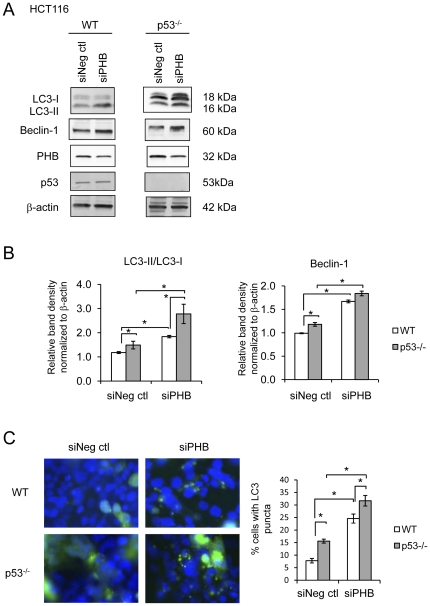
Autophagy is an adaptive response to extracellular and intracellular stress by which cytoplasmic components and organelles, including damaged mitochondria, are degraded to promote cell survival and restore cell homeostasis. Certain genes involved in autophagy confer susceptibility to Crohn's disease. Reactive oxygen species and pro-inflammatory cytokines such as tumor necrosis factor α (TNFα), both of which are increased during active inflammatory bowel disease, promote cellular injury and autophagy via mitochondrial damage. Prohibitin (PHB), which plays a role in maintaining normal mitochondrial respiratory function, is decreased during active inflammatory bowel disease. Restoration of colonic epithelial PHB expression protects mice from experimental colitis and combats oxidative stress. In this study, we investigated the potential role of PHB in modulating mitochondrial stress-related autophagy in intestinal epithelial cells.
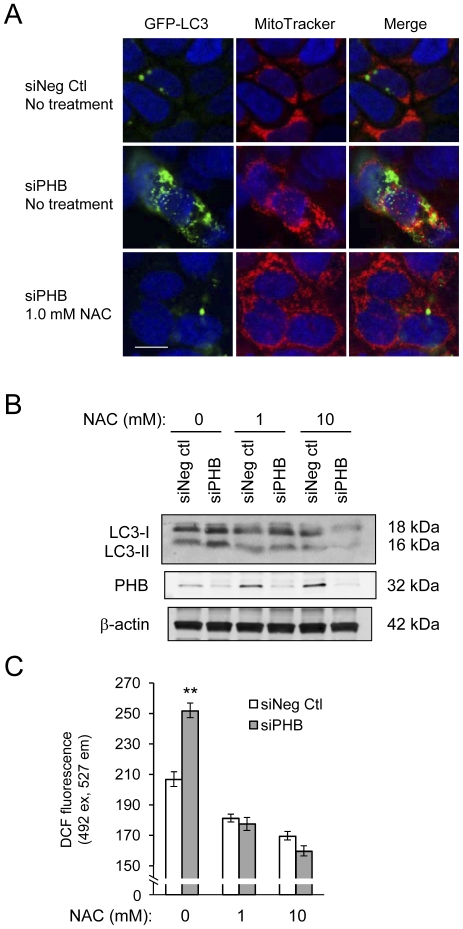
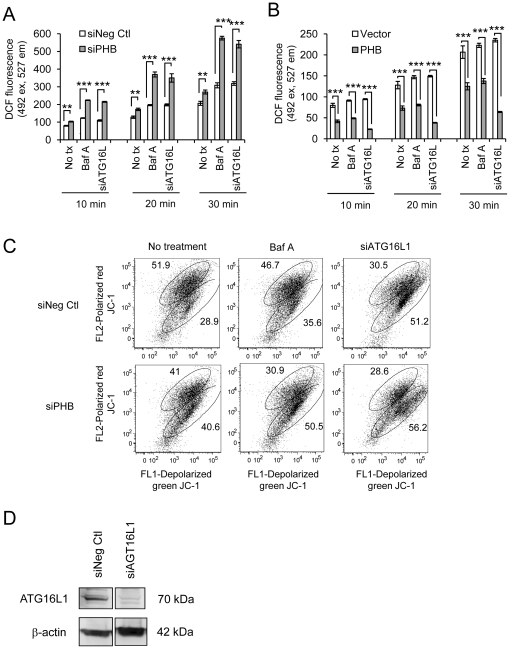
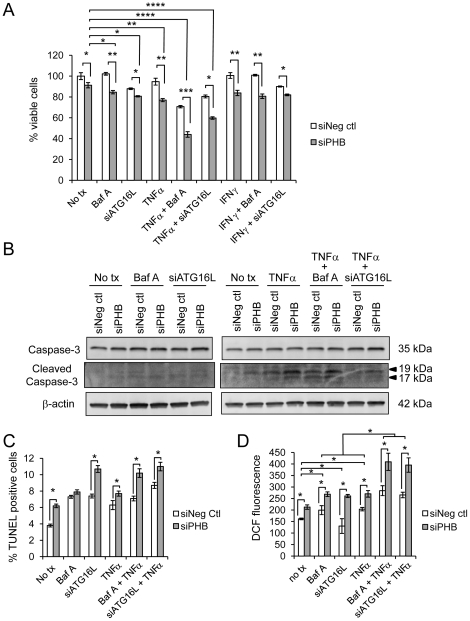
We measured autophagy activation in response to knockdown of PHB expression by RNA interference in Caco2-BBE and HCT116 WT and p53 null cells. The effect of exogenous PHB expression on TNFα- and IFNγ-induced autophagy was assessed. Autophagy was inhibited using Bafilomycin A(1) or siATG16L1 during PHB knockdown and the affect on intracellular oxidative stress, mitochondrial membrane potential, and cell viability were determined. The requirement of intracellular ROS in siPHB-induced autophagy was assessed using the ROS scavenger N-acetyl-L-cysteine.
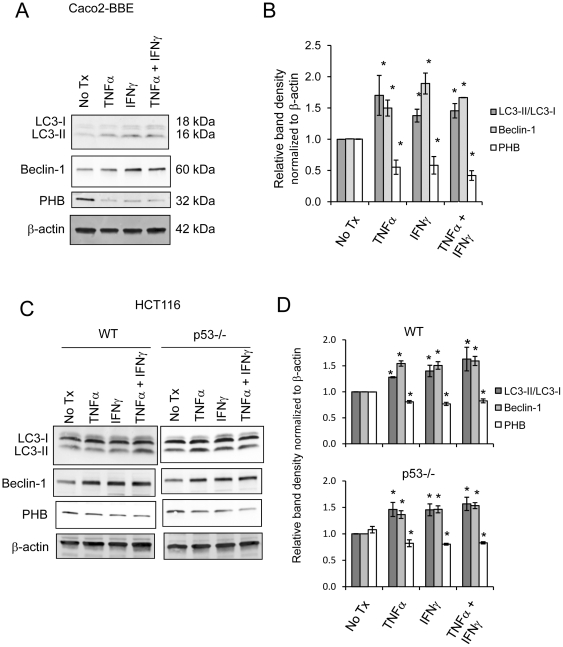
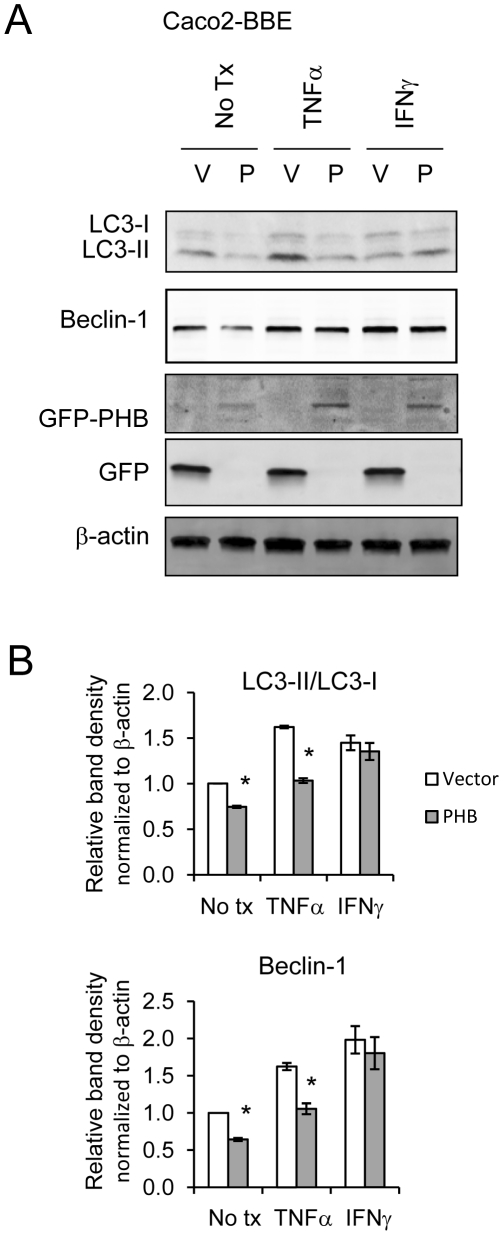
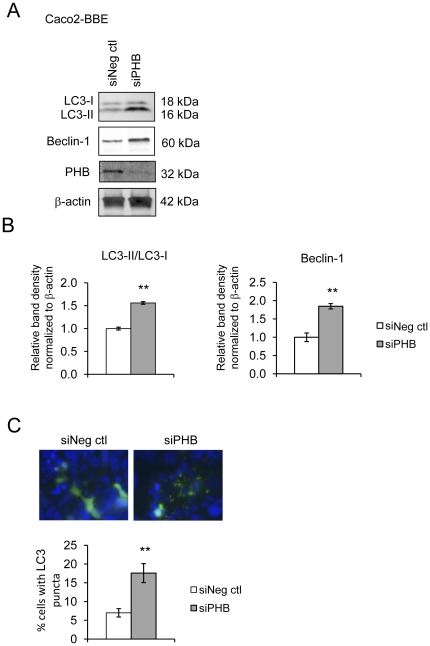
TNFα and IFNγ-induced autophagy inversely correlated with PHB protein expression. Exogenous PHB expression reduced basal autophagy and TNFα-induced autophagy. Gene silencing of PHB in epithelial cells induces mitochondrial autophagy via increased intracellular ROS. Inhibition of autophagy during PHB knockdown exacerbates mitochondrial depolarization and reduces cell viability.
Decreased PHB levels coupled with dysfunctional autophagy renders intestinal epithelial cells susceptible to mitochondrial damage and cytotoxicity. Repletion of PHB may represent a therapeutic approach to combat oxidant and cytokine-induced mitochondrial damage in diseases such as inflammatory bowel disease.
自噬是一种细胞内和细胞外应激的适应性反应,通过这种反应,细胞质成分和细胞器(包括受损的线粒体)被降解,以促进细胞存活并恢复细胞内稳态。某些参与自噬的基因易患克罗恩病。活性氧和促炎细胞因子(如肿瘤坏死因子-α(TNFα))在活动性炎症性肠病期间增加,通过线粒体损伤促进细胞损伤和自噬。在活动性炎症性肠病期间,发挥维持正常线粒体呼吸功能作用的抑制素(PHB)减少。结肠上皮 PHB 表达的恢复可保护小鼠免受实验性结肠炎的侵害,并对抗氧化应激。在这项研究中,我们研究了 PHB 在调节肠道上皮细胞中线粒体应激相关自噬中的潜在作用。
我们通过 RNA 干扰测量 PHB 表达敲低后 Caco2-BBE 和 HCT116 WT 和 p53 缺失细胞中自噬的激活。评估外源性 PHB 表达对 TNFα 和 IFNγ诱导的自噬的影响。在 PHB 敲低期间使用 Bafilomycin A(1)或 siATG16L1 抑制自噬,并测定细胞内氧化应激、线粒体膜电位和细胞活力的影响。使用 ROS 清除剂 N-乙酰-L-半胱氨酸评估 siPHB 诱导的自噬中细胞内 ROS 的必需性。
TNFα 和 IFNγ诱导的自噬与 PHB 蛋白表达呈负相关。外源性 PHB 表达降低了基础自噬和 TNFα 诱导的自噬。上皮细胞中 PHB 的基因沉默通过增加细胞内 ROS 诱导线粒体自噬。在 PHB 敲低期间抑制自噬会加剧线粒体去极化并降低细胞活力。
PHB 水平降低加上自噬功能障碍使肠道上皮细胞易受线粒体损伤和细胞毒性的影响。PHB 的补充可能代表一种治疗方法,可用于治疗炎症性肠病等疾病中的氧化剂和细胞因子诱导的线粒体损伤。