Chen Han, Hendricks Audrey E, Cheng Yansong, Cupples Adrienne L, Dupuis Josée, Liu Ching-Ti
Department of Biostatistics, Boston University School of Public Health, Boston, MA 02118, USA.
BMC Proc. 2011 Nov 29;5 Suppl 9(Suppl 9):S113. doi: 10.1186/1753-6561-5-S9-S113.
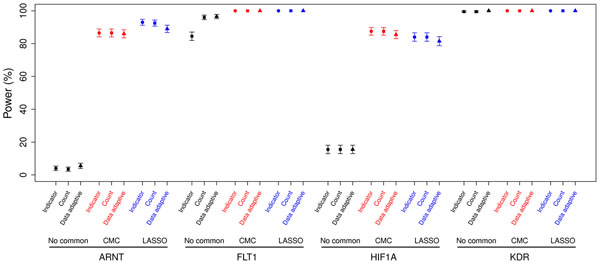
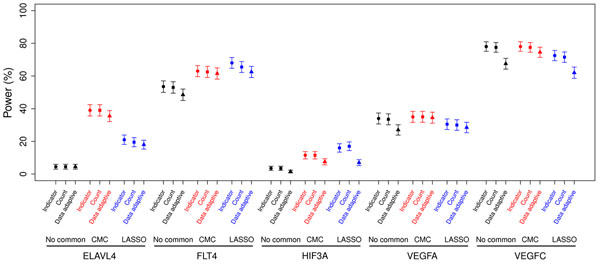
With recent advances in technology, deep sequencing data will be widely used to further the understanding of genetic influence on traits of interest. Therefore not only common variants but also rare variants need to be better used to exploit the new information provided by deep sequencing data. Recently, statistical approaches for analyzing rare variants in genetic association studies have been proposed, but many of them were designed only for dichotomous outcomes. We compare the type I error and power of several statistical approaches applicable to quantitative traits for collapsing and analyzing rare variant data within a defined gene region. In addition to comparing methods that consider only rare variants, such as indicator, count, and data-adaptive collapsing methods, we also compare methods that incorporate the analysis of common variants along with rare variants, such as CMC and LASSO regression. We find that the three methods used to collapse rare variants perform similarly in this simulation setting where all risk variants were simulated to have effects in the same direction. Further, we find that incorporating common variants is beneficial and using a LASSO regression to choose which common variants to include is most useful when there is are few common risk variants compared to the total number of risk variants.
随着技术的最新进展,深度测序数据将被广泛用于进一步理解基因对感兴趣性状的影响。因此,不仅常见变异,稀有变异也需要得到更好的利用,以挖掘深度测序数据提供的新信息。最近,已经提出了在基因关联研究中分析稀有变异的统计方法,但其中许多方法仅针对二分结局设计。我们比较了几种适用于定量性状的统计方法在定义的基因区域内汇总和分析稀有变异数据时的I型错误率和检验效能。除了比较仅考虑稀有变异的方法,如指示法、计数法和数据自适应汇总法外,我们还比较了将常见变异分析与稀有变异分析相结合的方法,如CMC和LASSO回归。我们发现,在所有风险变异被模拟为具有相同方向效应的这种模拟设置中,用于汇总稀有变异的三种方法表现相似。此外,我们发现纳入常见变异是有益的,并且当与风险变异总数相比常见风险变异较少时,使用LASSO回归来选择纳入哪些常见变异最为有用。