Ao Zhimin, Jiang Man, Wen Zi, Li Sean
Key Laboratory of Automobile Materials, Ministry of Education and Department of Materials Science and Engineering, Jilin University, Changchun, 130022, China.
Nanoscale Res Lett. 2012 Feb 28;7(1):158. doi: 10.1186/1556-276X-7-158.
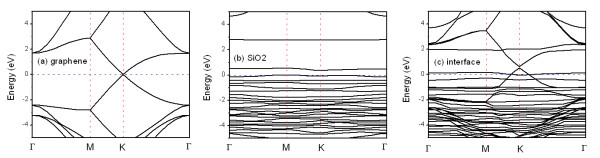
In this work, the graphene/α-SiO2(0001) interface is calculated using density functional theory. On the oxygen-terminated SiO2 surface, atomic structure reconstruction occurs at the graphene/SiO2 interface to eliminate the dangling bonds. The interface interaction is 77 meV/C atom, which indicates that van der Waals force dominates the interaction, but it is stronger than the force between the graphene layers in graphite. The distance between graphene and the SiO2 surface is 2.805 Å, which is smaller than the 3.4 Å interlayer distance of graphite. In addition, the SiO2 substrate induces p-type doping in graphene and opens a small gap of 0.13 eV at the Dirac point of graphene, which is desirable for electronic device applications.
在本工作中,利用密度泛函理论计算了石墨烯/α-SiO2(0001)界面。在氧端终止的SiO2表面,石墨烯/SiO2界面处发生原子结构重构以消除悬空键。界面相互作用为77 meV/C原子,这表明范德华力主导该相互作用,但它比石墨中石墨烯层之间的力更强。石墨烯与SiO2表面之间的距离为2.805 Å,小于石墨的3.4 Å层间距。此外,SiO2衬底在石墨烯中诱导p型掺杂,并在石墨烯的狄拉克点处打开一个0.13 eV的小能隙,这对于电子器件应用是有利的。