Petersen Ashley, Sitarik Alexandra, Luedtke Alexander, Powers Scott, Bekmetjev Airat, Tintle Nathan L
Departments of Mathematics, Computer Science, and Statistics, St. Olaf College, 1520 St. Olaf Avenue, Northfield, MN 55057, USA.
Department of Mathematics, Wittenberg University, 200 West Ward Street, Springfield, OH 45501, USA.
BMC Proc. 2011 Nov 29;5 Suppl 9(Suppl 9):S48. doi: 10.1186/1753-6561-5-S9-S48.
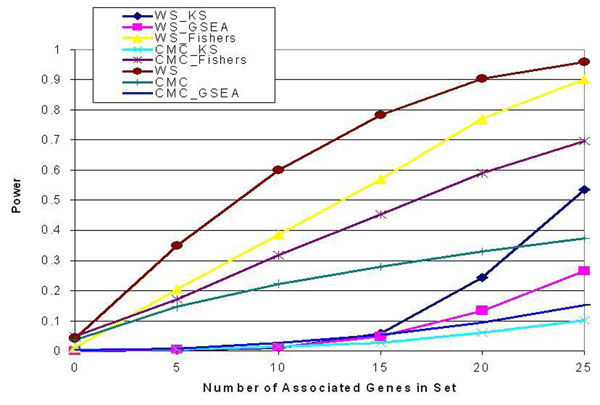
Analyzing sets of genes in genome-wide association studies is a relatively new approach that aims to capitalize on biological knowledge about the interactions of genes in biological pathways. This approach, called pathway analysis or gene set analysis, has not yet been applied to the analysis of rare variants. Applying pathway analysis to rare variants offers two competing approaches. In the first approach rare variant statistics are used to generate p-values for each gene (e.g., combined multivariate collapsing [CMC] or weighted-sum [WS]) and the gene-level p-values are combined using standard pathway analysis methods (e.g., gene set enrichment analysis or Fisher's combined probability method). In the second approach, rare variant methods (e.g., CMC and WS) are applied directly to sets of single-nucleotide polymorphisms (SNPs) representing all SNPs within genes in a pathway. In this paper we use simulated phenotype and real next-generation sequencing data from Genetic Analysis Workshop 17 to analyze sets of rare variants using these two competing approaches. The initial results suggest substantial differences in the methods, with Fisher's combined probability method and the direct application of the WS method yielding the best power. Evidence suggests that the WS method works well in most situations, although Fisher's method was more likely to be optimal when the number of causal SNPs in the set was low but the risk of the causal SNPs was high.
在全基因组关联研究中分析基因集是一种相对较新的方法,其目的是利用有关生物途径中基因相互作用的生物学知识。这种方法,称为途径分析或基因集分析,尚未应用于罕见变异的分析。将途径分析应用于罕见变异提供了两种相互竞争的方法。在第一种方法中,使用罕见变异统计为每个基因生成p值(例如,联合多变量合并[CMC]或加权和[WS]),并使用标准途径分析方法(例如,基因集富集分析或Fisher联合概率法)合并基因水平的p值。在第二种方法中,将罕见变异方法(例如,CMC和WS)直接应用于代表途径中基因内所有单核苷酸多态性(SNP)的SNP集。在本文中,我们使用来自遗传分析研讨会17的模拟表型和真实下一代测序数据,通过这两种相互竞争的方法分析罕见变异集。初步结果表明这些方法存在实质性差异,Fisher联合概率法和WS方法的直接应用产生了最佳效能。有证据表明,WS方法在大多数情况下效果良好,尽管当集合中因果SNP的数量较低但因果SNP的风险较高时,Fisher方法更有可能是最优的。