AIDS Research Institute IrsiCaixa, Hospital Universitari Germans Trias i Pujol, Badalona, Barcelona, Spain.
PLoS One. 2012;7(2):e32714. doi: 10.1371/journal.pone.0032714. Epub 2012 Feb 29.
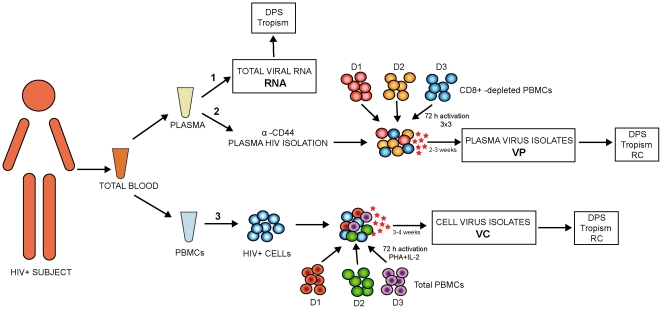
The use of in vitro models to unravel the phenotypic characteristics of circulating viral variants is key to understanding HIV-1 pathogenesis but limited by the availability of primary viral isolates from biological samples. However, overall in vivo genetic variability of HIV-1 within a subject may not be reflected in the viable viral population obtained after isolation. Although several studies have tried to determine whether viral populations expanded in vitro are representative of in vivo findings, the answer remains unclear due to the reduced number of clonal sequences analyzed or samples compared. In order to overcome previous experimental limitations, here we applied Deep Pyrosequencing (DPS) technology in combination with phenotypic experiments to analyze and compare with unprecedented detail the composition of viral isolates and in vivo quasispecies.
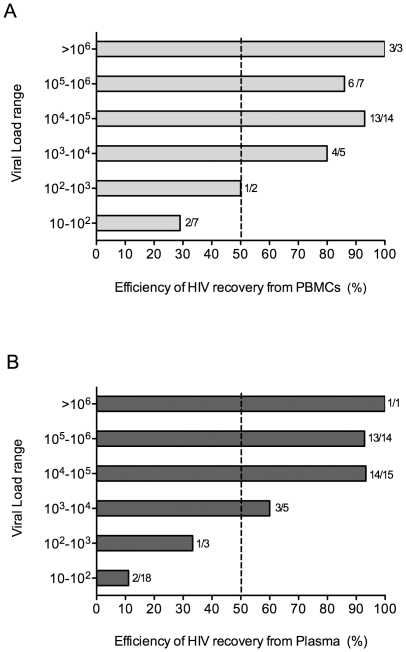
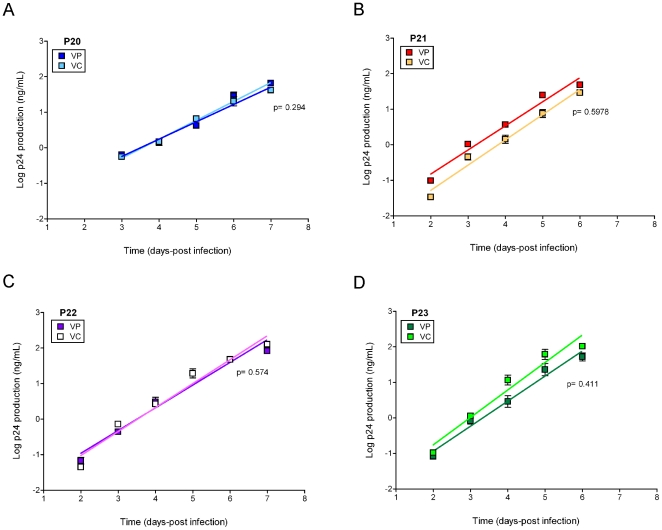
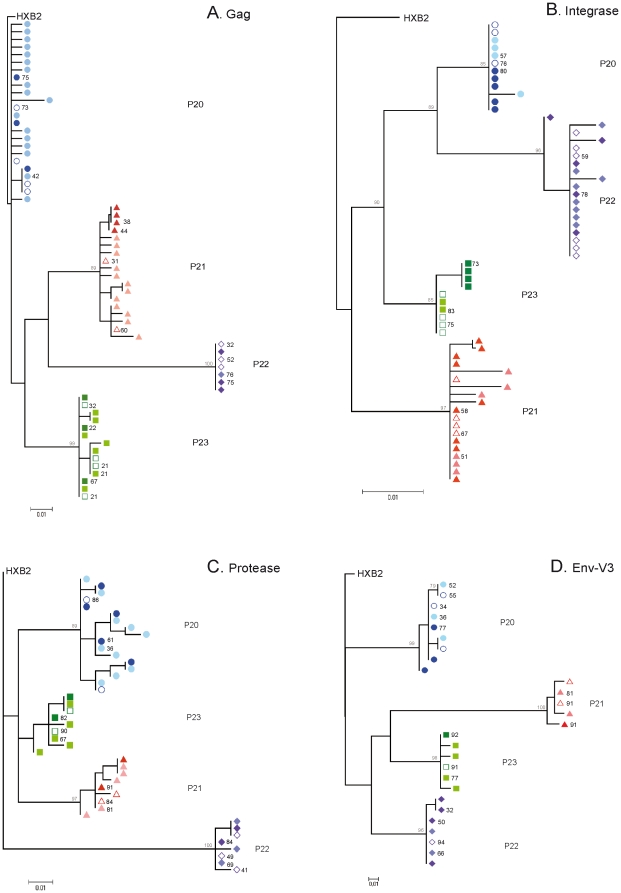
METHODOLOGY/PRINCIPAL FINDINGS: We amplified by DPS HIV-1 genomic regions covering gag, protease, integrase and env-V3 to characterize paired isolates from plasma and peripheral blood mononuclear cells and compare them with total plasma viral RNA in four recently HIV-1 infected subjects. Our study demonstrated the presence of unique haplotypes scattered between sample types with conservation of major variants. In addition, no differences in intra- and inter-population encoded protein variability were found between the different types of isolates or when these were compared to plasma viral RNA within subjects. Additionally, in vitro experiments demonstrated phenotypic similarities in terms of replicative capacity and co-receptor usage between viral isolates and plasma viral RNA.
This study is the first in-depth comparison and characterization of viral isolates from different sources and plasma circulating quasispecies using DPS in recently HIV-1 infected subjects. Our data supports the use of primary isolates regardless of their plasma or cellular origin to define genetic variability and biological traits of circulating HIV-1 quasispecies.
利用体外模型来揭示循环病毒变异体的表型特征是理解 HIV-1 发病机制的关键,但受到从生物样本中获得原发性病毒分离物的限制。然而,HIV-1 在个体体内的总体遗传变异性可能无法反映在分离后获得的可培养病毒群体中。尽管有几项研究试图确定在体外扩增的病毒群体是否代表体内发现,但由于分析或比较的克隆序列数量较少,答案仍不清楚。为了克服以前的实验局限性,我们在这里应用深度焦磷酸测序(DPS)技术结合表型实验,以前所未有的细节分析和比较病毒分离物和体内准种的组成。
方法/主要发现:我们通过 DPS 扩增了涵盖 gag、蛋白酶、整合酶和 env-V3 的 HIV-1 基因组区域,以表征来自血浆和外周血单核细胞的配对分离物,并将其与四个最近感染 HIV-1 的个体的总血浆病毒 RNA 进行比较。我们的研究表明,独特的单倍型存在于样本类型之间,主要变体保持不变。此外,在不同类型的分离物之间或在个体内将这些分离物与血浆病毒 RNA 进行比较时,未发现种群内和种群间编码蛋白变异性的差异。此外,体外实验表明,病毒分离物和血浆病毒 RNA 在复制能力和共受体使用方面具有表型相似性。
这是首次在最近感染 HIV-1 的个体中使用 DPS 对来自不同来源的病毒分离物和血浆循环准种进行深入比较和特征描述。我们的数据支持使用原发性分离物,无论其来自血浆还是细胞,以定义循环 HIV-1 准种的遗传变异性和生物学特征。