Instituto de Bioquímica Médica, Universidade Federal do Rio de Janeiro, Cidade Universitária, Rio de Janeiro, Rio de Janeiro, Brazil.
PLoS One. 2012;7(4):e33871. doi: 10.1371/journal.pone.0033871. Epub 2012 Apr 2.
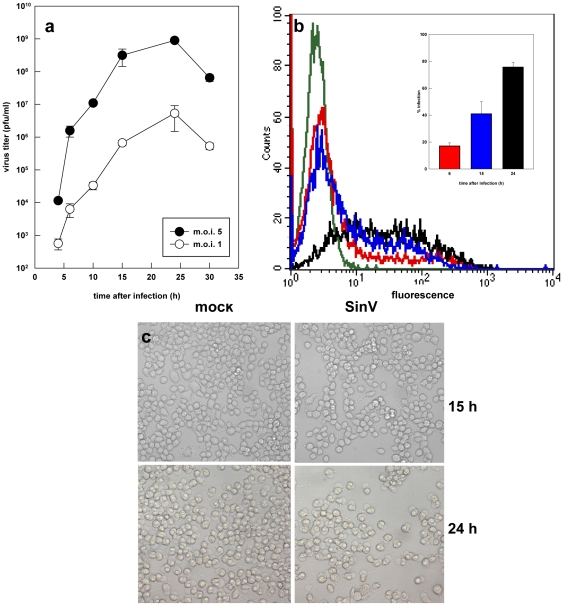
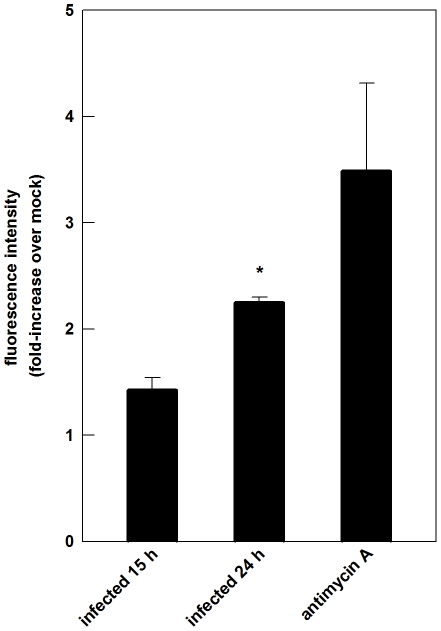
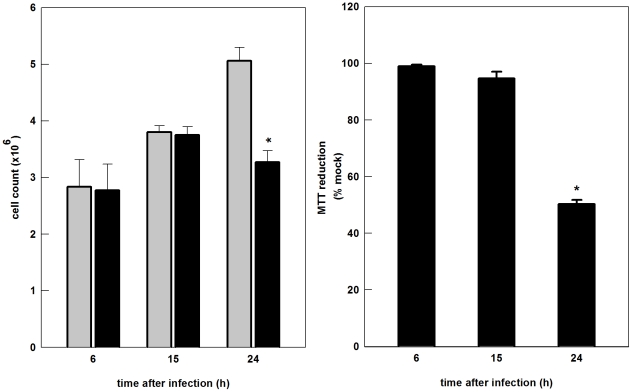
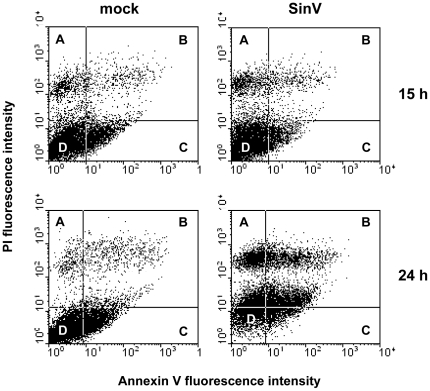
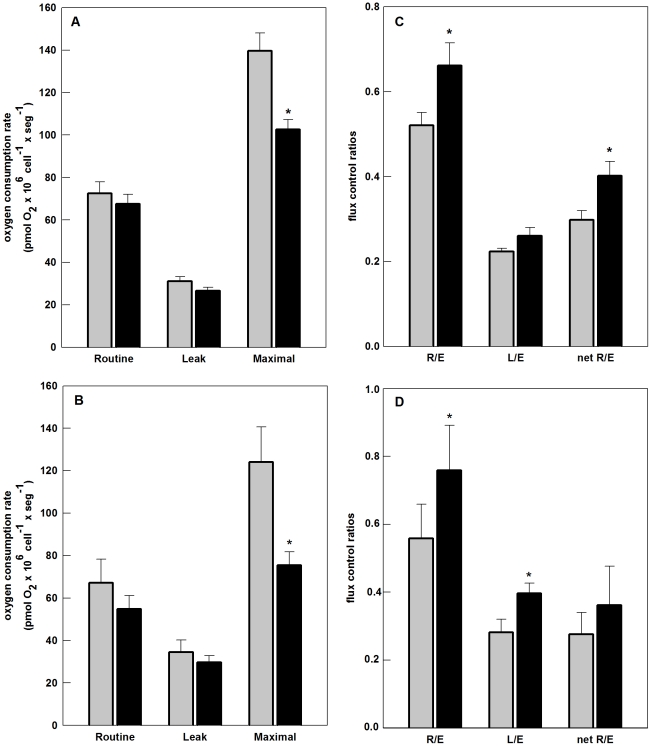
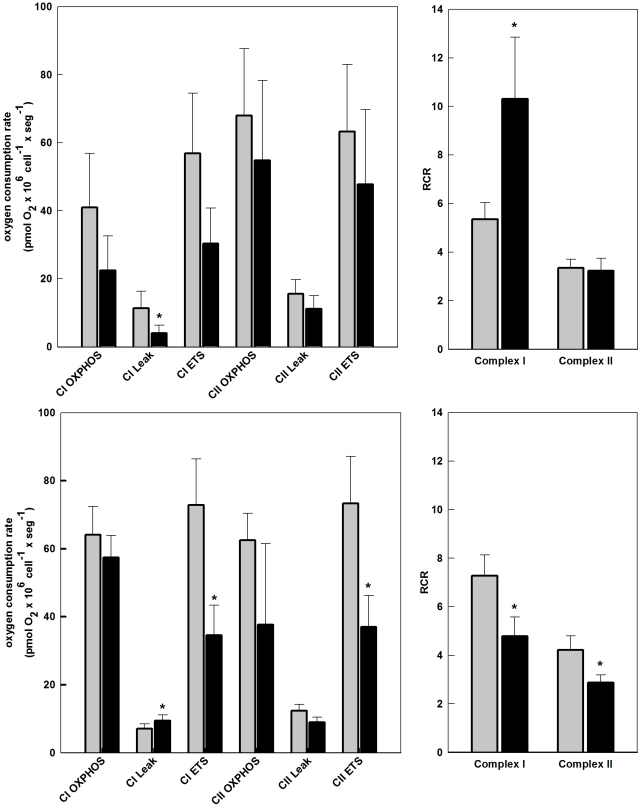
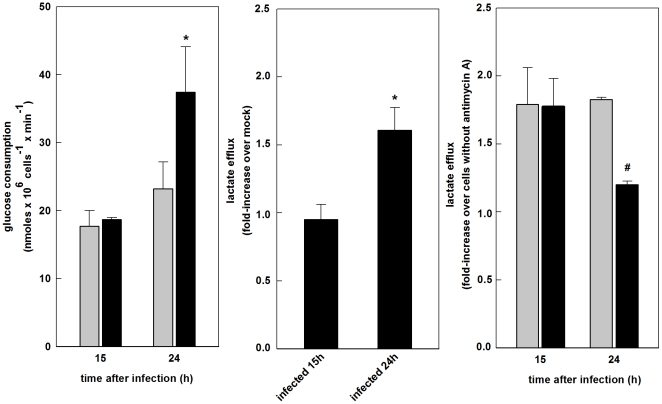
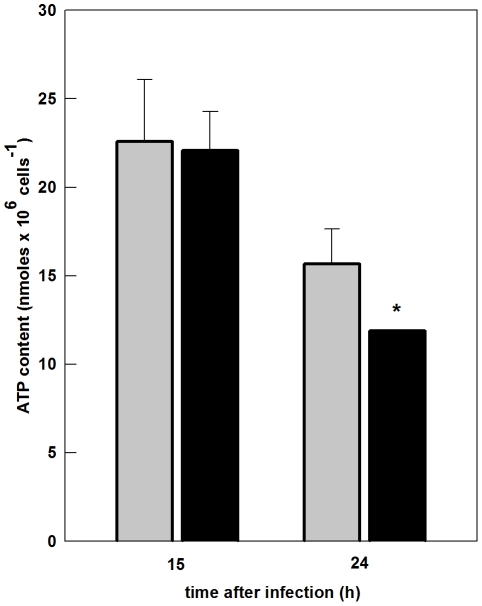
The metabolic resources crucial for viral replication are provided by the host. Details of the mechanisms by which viruses interact with host metabolism, altering and recruiting high free-energy molecules for their own replication, remain unknown. Sindbis virus, the prototype of and most widespread alphavirus, causes outbreaks of arthritis in humans and serves as a model for the study of the pathogenesis of neurological diseases induced by alphaviruses in mice. In this work, respirometric analysis was used to evaluate the effects of Sindbis virus infection on mitochondrial bioenergetics of a mouse neuroblastoma cell lineage, Neuro 2a. The modulation of mitochondrial functions affected cellular ATP content and this was synchronous with Sindbis virus replication cycle and cell death. At 15 h, irrespective of effects on cell viability, viral replication induced a decrease in oxygen consumption uncoupled to ATP synthesis and a 36% decrease in maximum uncoupled respiration, which led to an increase of 30% in the fraction of oxygen consumption used for ATP synthesis. Decreased proton leak associated to complex I respiration contributed to the apparent improvement of mitochondrial function. Cellular ATP content was not affected by infection. After 24 h, mitochondria dysfunction was clearly observed as maximum uncoupled respiration reduced 65%, along with a decrease in the fraction of oxygen consumption used for ATP synthesis. Suppressed respiration driven by complexes I- and II-related substrates seemed to play a role in mitochondrial dysfunction. Despite the increase in glucose uptake and glycolytic flux, these changes were followed by a 30% decrease in ATP content and neuronal death. Taken together, mitochondrial bioenergetics is modulated during Sindbis virus infection in such a way as to favor ATP synthesis required to support active viral replication. These early changes in metabolism of Neuro 2a cells may form the molecular basis of neuronal dysfunction and Sindbis virus-induced encephalitis.
病毒复制所需的代谢资源由宿主提供。病毒与宿主代谢相互作用的机制细节,以及改变和招募高能量分子用于自身复制的机制仍不清楚。辛德毕斯病毒是甲病毒的原型和最广泛的病毒,它会导致人类关节炎爆发,并作为研究甲病毒在小鼠中引起的神经疾病发病机制的模型。在这项工作中,使用呼吸计分析来评估辛德毕斯病毒感染对小鼠神经母细胞瘤系 Neuro 2a 的线粒体生物能量学的影响。受感染细胞中线粒体功能的调节会影响细胞内的 ATP 含量,并且与辛德毕斯病毒的复制周期和细胞死亡同步。在 15 小时,无论对细胞活力的影响如何,病毒复制都会导致与 ATP 合成解耦的耗氧量减少和最大解耦呼吸减少 36%,从而导致用于 ATP 合成的耗氧量增加 30%。与复合物 I 呼吸相关的质子泄漏减少有助于改善线粒体功能的表观。细胞内的 ATP 含量不受感染影响。在 24 小时后,线粒体功能障碍明显,最大解耦呼吸减少 65%,同时用于 ATP 合成的耗氧量减少。复合物 I-和 II-相关底物驱动的受抑制呼吸似乎在线粒体功能障碍中发挥作用。尽管葡萄糖摄取和糖酵解通量增加,但这些变化后细胞内的 ATP 含量下降 30%,并导致神经元死亡。总之,在辛德毕斯病毒感染过程中,线粒体生物能量学被调节,以有利于支持活跃病毒复制所需的 ATP 合成。Neuro 2a 细胞代谢的这些早期变化可能构成神经元功能障碍和辛德毕斯病毒引起的脑炎的分子基础。