Committee on Evolutionary Biology, University of Chicago, Chicago, Illinois, United States of America.
PLoS One. 2012;7(4):e33394. doi: 10.1371/journal.pone.0033394. Epub 2012 Apr 6.
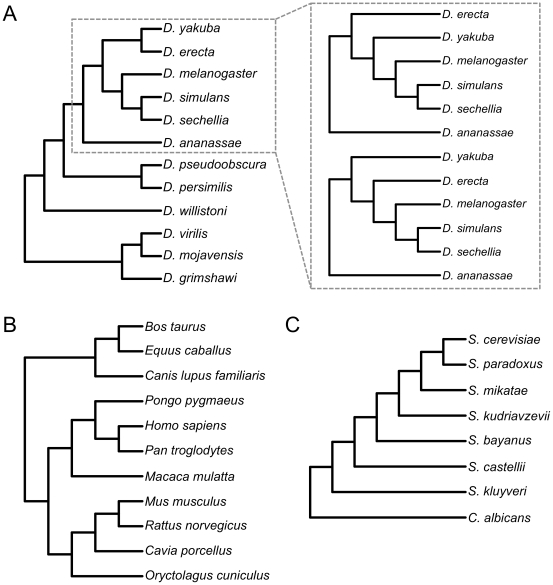
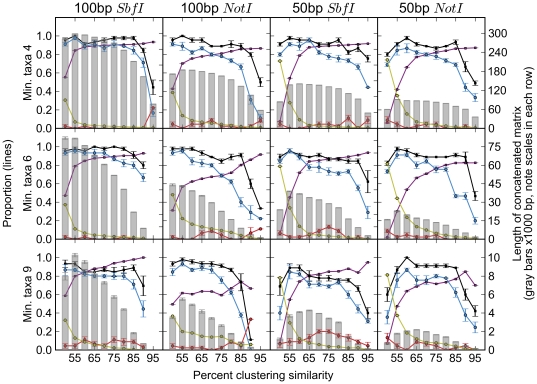
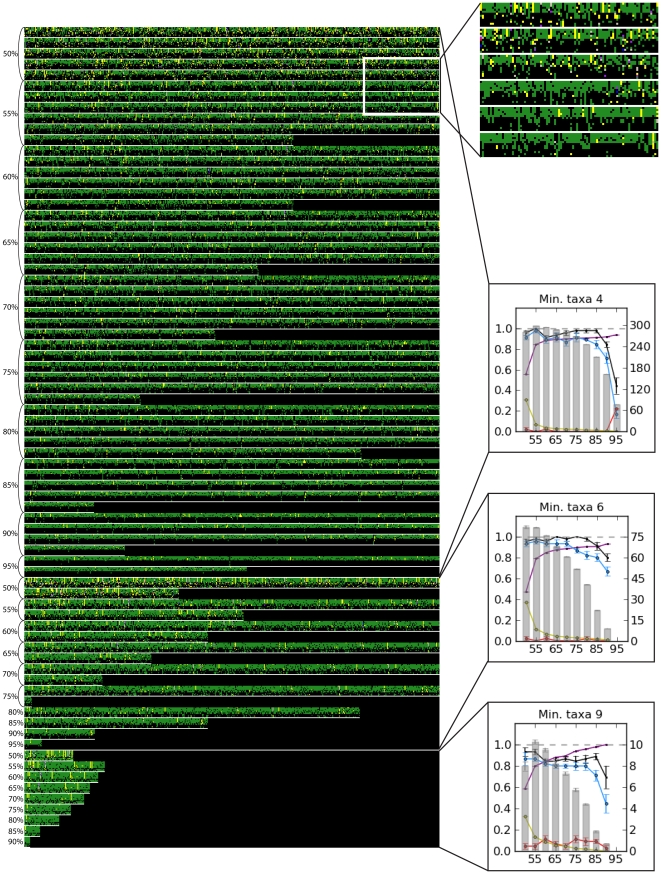
Reduced-representation genome sequencing represents a new source of data for systematics, and its potential utility in interspecific phylogeny reconstruction has not yet been explored. One approach that seems especially promising is the use of inexpensive short-read technologies (e.g., Illumina, SOLiD) to sequence restriction-site associated DNA (RAD)--the regions of the genome that flank the recognition sites of restriction enzymes. In this study, we simulated the collection of RAD sequences from sequenced genomes of different taxa (Drosophila, mammals, and yeasts) and developed a proof-of-concept workflow to test whether informative data could be extracted and used to accurately reconstruct "known" phylogenies of species within each group. The workflow consists of three basic steps: first, sequences are clustered by similarity to estimate orthology; second, clusters are filtered by taxonomic coverage; and third, they are aligned and concatenated for "total evidence" phylogenetic analysis. We evaluated the performance of clustering and filtering parameters by comparing the resulting topologies with well-supported reference trees and we were able to identify conditions under which the reference tree was inferred with high support. For Drosophila, whole genome alignments allowed us to directly evaluate which parameters most consistently recovered orthologous sequences. For the parameter ranges explored, we recovered the best results at the low ends of sequence similarity and taxonomic representation of loci; these generated the largest supermatrices with the highest proportion of missing data. Applications of the method to mammals and yeasts were less successful, which we suggest may be due partly to their much deeper evolutionary divergence times compared to Drosophila (crown ages of approximately 100 and 300 versus 60 Mya, respectively). RAD sequences thus appear to hold promise for reconstructing phylogenetic relationships in younger clades in which sufficient numbers of orthologous restriction sites are retained across species.
简化基因组测序为系统发生学提供了新的数据来源,但其在种间系统发育重建中的潜在应用尚未得到探索。一种似乎很有前途的方法是利用廉价的短读长技术(例如 Illumina、SOLiD)对基因组侧翼限制酶识别位点的限制相关 DNA(RAD)区域进行测序。在这项研究中,我们模拟了来自不同分类群(果蝇、哺乳动物和酵母)测序基因组的 RAD 序列的采集,并开发了一个概念验证工作流程,以测试是否可以提取有用的数据并用于准确重建每个组内“已知”物种的系统发育。该工作流程由三个基本步骤组成:首先,通过相似性聚类来估计同源性;其次,根据分类覆盖范围过滤聚类;最后,对它们进行对齐和串联,用于“全证据”系统发育分析。我们通过将得到的拓扑结构与具有良好支持的参考树进行比较,评估了聚类和过滤参数的性能,并且能够确定在哪些条件下可以用高支持度推断出参考树。对于果蝇,全基因组比对使我们能够直接评估哪些参数最一致地恢复了同源序列。在所探索的参数范围内,我们在序列相似性和基因座分类代表的低端恢复了最佳结果;这些结果生成了最大的超级矩阵,具有最高比例的缺失数据。该方法在哺乳动物和酵母中的应用效果较差,我们认为这可能部分是由于它们与果蝇相比进化分歧时间更长(大约 100 和 300 与 60 Mya,分别为冠年龄)。因此,RAD 序列似乎有望在保留足够数量的物种间同源限制位点的年轻进化枝中重建系统发育关系。