Seetharam Arun, Stuart Gary W
Department of Biology, Indiana State University, Terre Haute, Indiana 47809, USA.
BMC Res Notes. 2012 Dec 4;5:670. doi: 10.1186/1756-0500-5-670.
Reconstructing the evolutionary history of organisms using traditional phylogenetic methods may suffer from inaccurate sequence alignment. An alternative approach, particularly effective when whole genome sequences are available, is to employ methods that don't use explicit sequence alignments. We extend a novel phylogenetic method based on Singular Value Decomposition (SVD) to reconstruct the phylogeny of 12 sequenced Drosophila species. SVD analysis provides accurate comparisons for a high fraction of sequences within whole genomes without the prior identification of orthologs or homologous sites. With this method all protein sequences are converted to peptide frequency vectors within a matrix that is decomposed to provide simplified vector representations for each protein of the genome in a reduced dimensional space. These vectors are summed together to provide a vector representation for each species, and the angle between these vectors provides distance measures that are used to construct species trees.
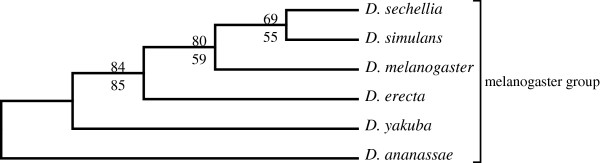
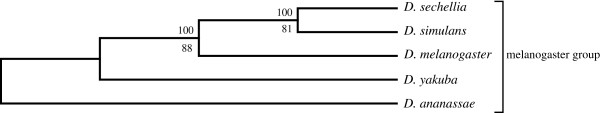
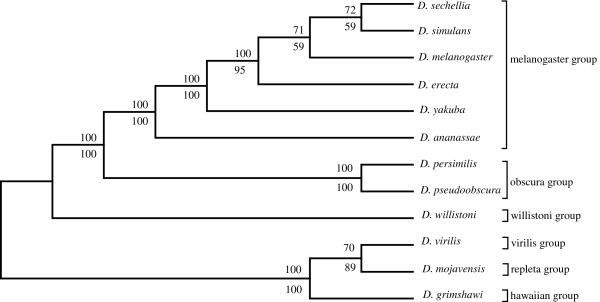
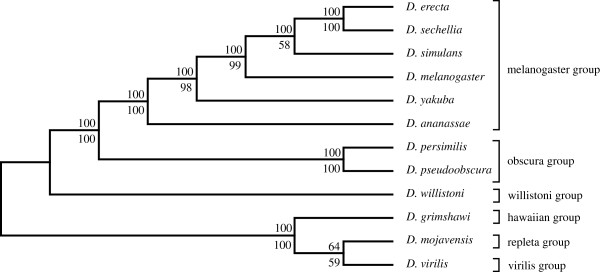
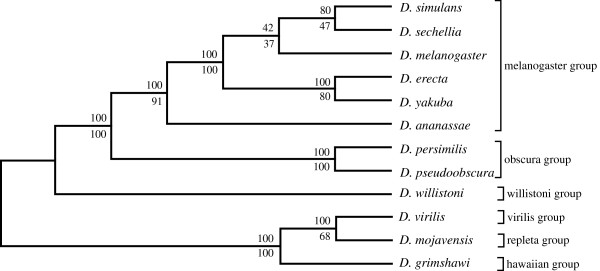
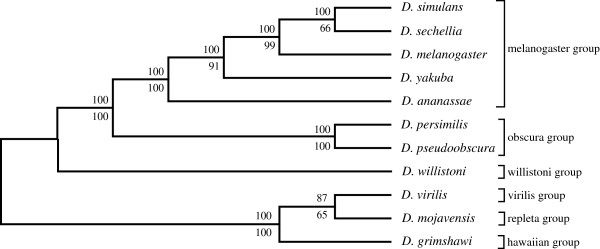
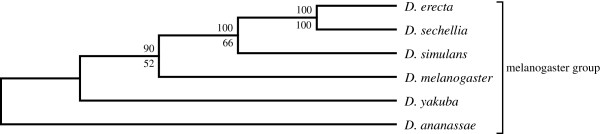
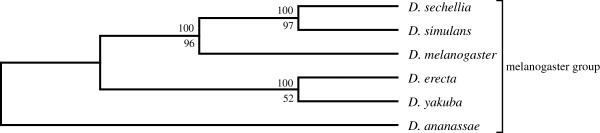
An unfiltered whole genome analysis (193,622 predicted proteins) strongly supports the currently accepted phylogeny for 12 Drosophila species at higher dimensions except for the generally accepted but difficult to discern sister relationship between D. erecta and D. yakuba. Also, in accordance with previous studies, many sequences appear to support alternative phylogenies. In this case, we observed grouping of D. erecta with D. sechellia when approximately 55% to 95% of the proteins were removed using a filter based on projection values or by reducing resolution by using fewer dimensions. Similar results were obtained when just the melanogaster subgroup was analyzed.
These results indicate that using our novel phylogenetic method, it is possible to consult and interpret all predicted protein sequences within multiple whole genomes to produce accurate phylogenetic estimations of relatedness between Drosophila species. Furthermore, protein filtering can be effectively applied to reduce incongruence in the dataset as well as to generate alternative phylogenies.
使用传统系统发育方法重建生物体的进化历史可能会受到不准确序列比对的影响。另一种方法,尤其是在有全基因组序列时特别有效,是采用不使用显式序列比对的方法。我们扩展了一种基于奇异值分解(SVD)的新型系统发育方法,以重建12个已测序果蝇物种的系统发育。SVD分析可在无需事先鉴定直系同源物或同源位点的情况下,对全基因组内的大部分序列进行准确比较。使用这种方法,所有蛋白质序列都被转换为矩阵内的肽频率向量,该矩阵被分解以在降维空间中为基因组中的每个蛋白质提供简化的向量表示。这些向量被加在一起,为每个物种提供一个向量表示,这些向量之间的夹角提供用于构建物种树的距离度量。
未经过滤的全基因组分析(193,622个预测蛋白质)在较高维度上有力地支持了目前被接受的12个果蝇物种的系统发育,除了公认但难以区分的直翅果蝇和雅库布果蝇之间的姐妹关系。此外,与先前的研究一致,许多序列似乎支持替代系统发育。在这种情况下,当使用基于投影值的过滤器或通过使用较少维度降低分辨率去除约55%至95%的蛋白质时,我们观察到直翅果蝇与塞舌尔果蝇聚在一起。仅分析黑腹果蝇亚组时也获得了类似结果。
这些结果表明,使用我们的新型系统发育方法,可以查阅和解释多个全基因组内的所有预测蛋白质序列,以对果蝇物种之间的亲缘关系进行准确的系统发育估计。此外,蛋白质过滤可以有效地应用于减少数据集中的不一致性,并生成替代系统发育。