Herrera Santiago, Reyes-Herrera Paula H, Shank Timothy M
Biology Department, Woods Hole Oceanographic Institution Biology Department, Massachusetts Institute of Technology
Colombian Corporation for Agricultural Research (CORPOICA), Bogotá, Colombia.
Genome Biol Evol. 2015 Nov 3;7(12):3207-25. doi: 10.1093/gbe/evv210.
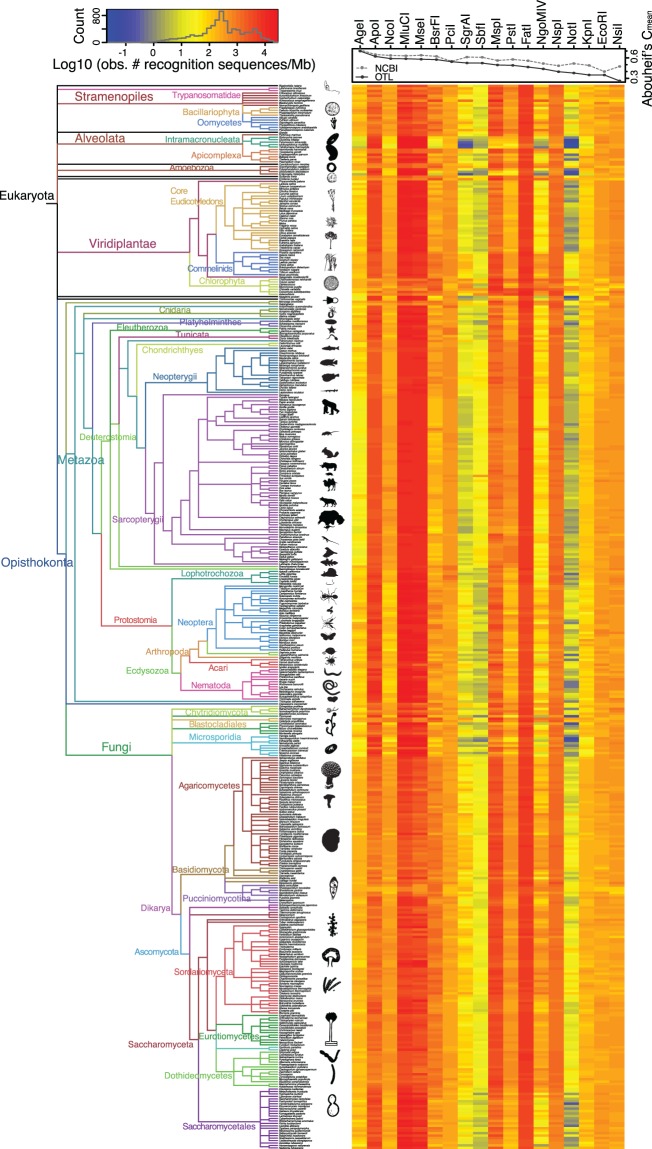
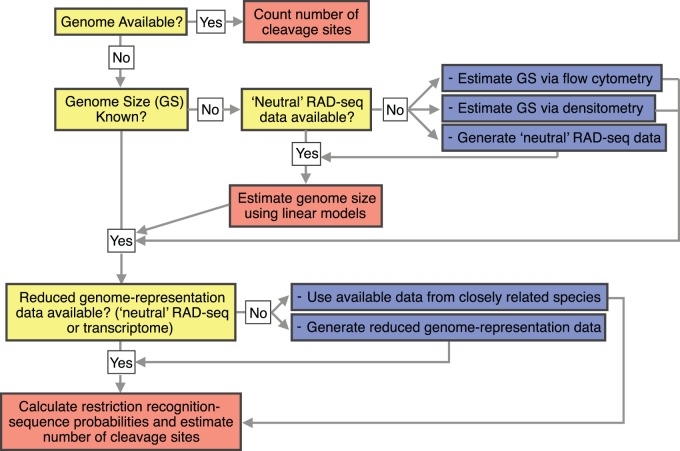
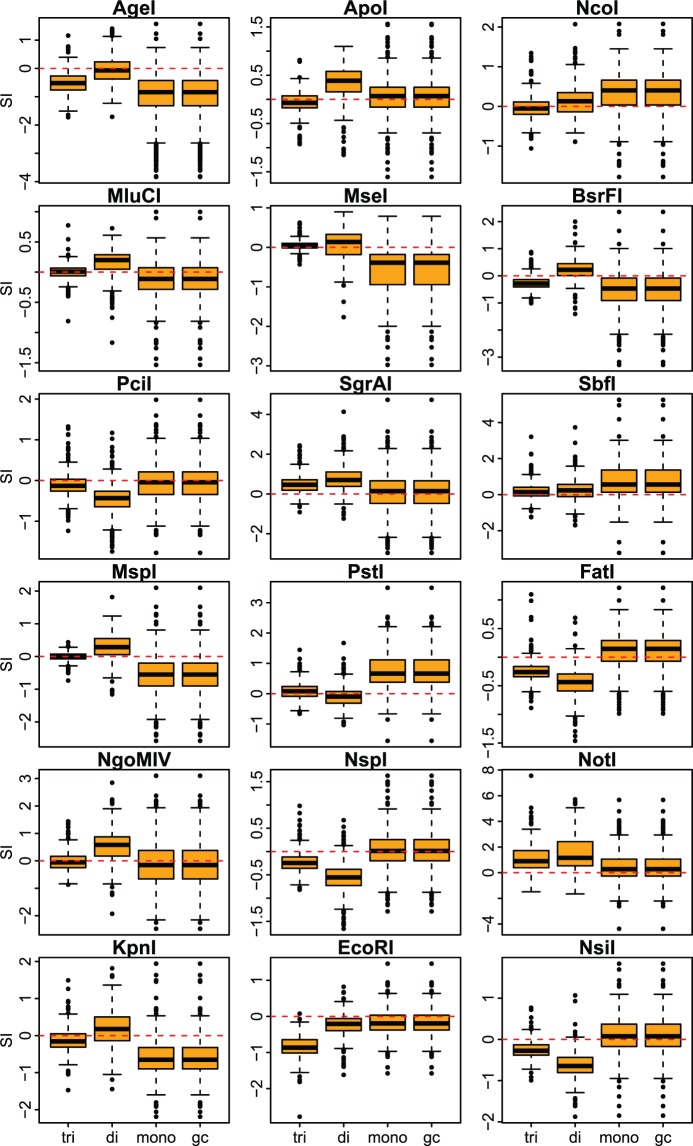
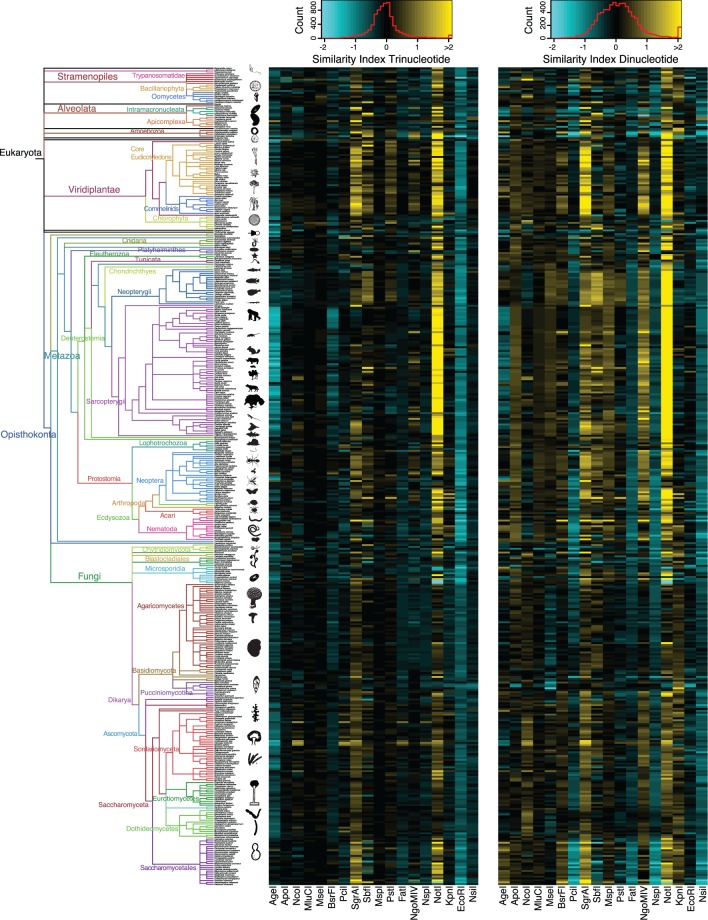
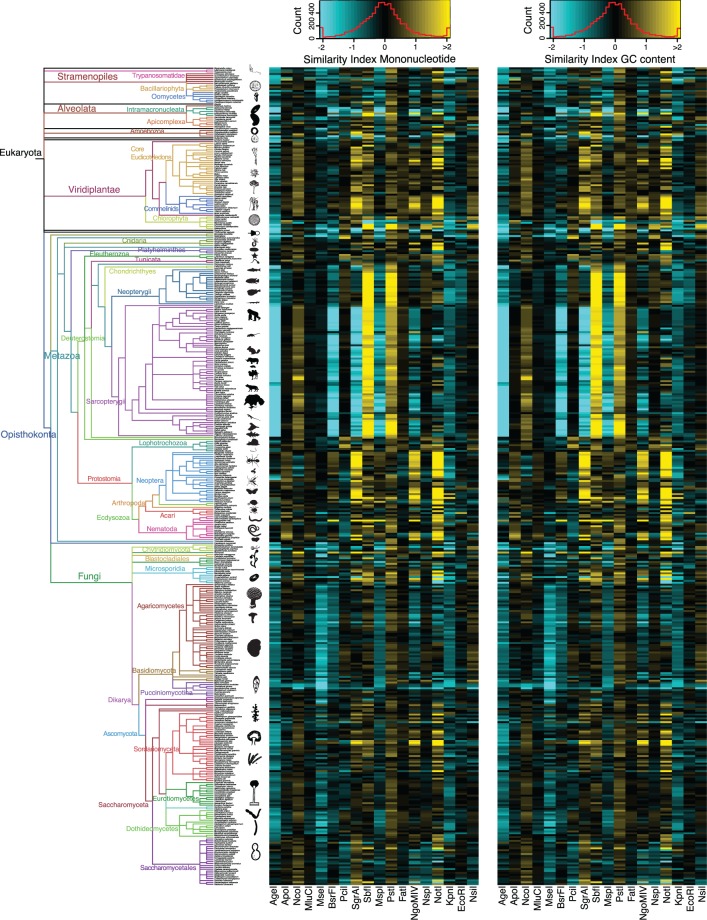
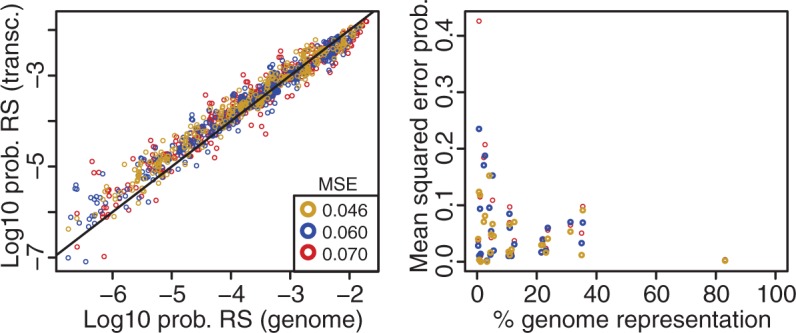
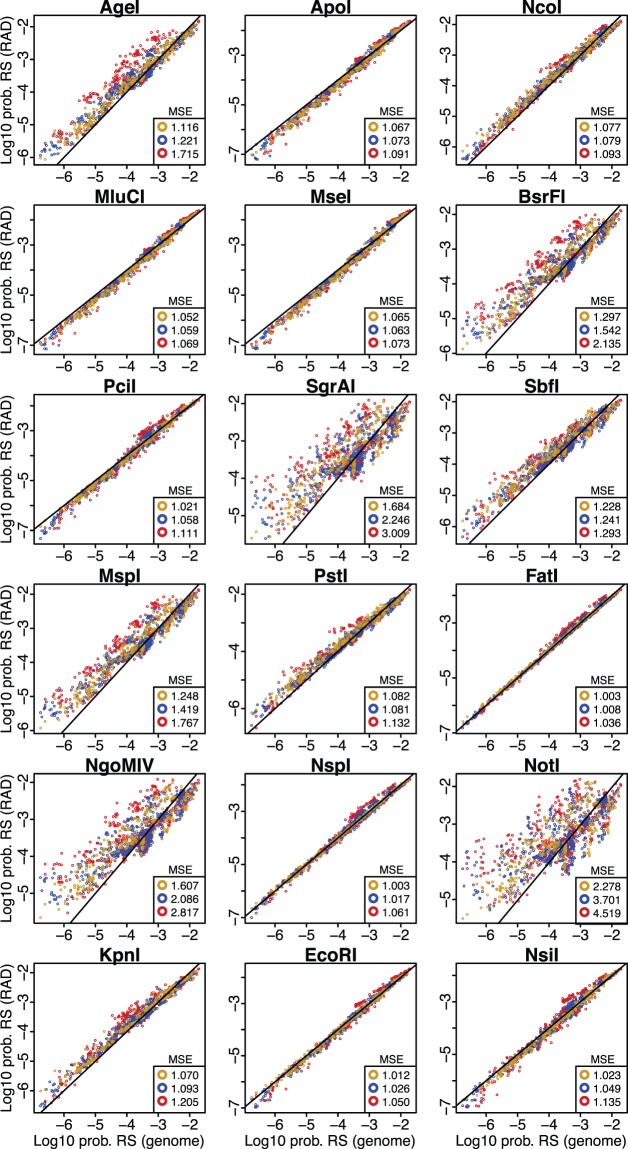
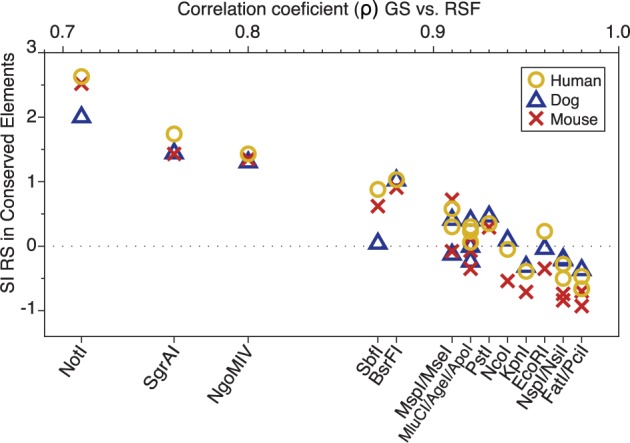
High-throughput sequencing of reduced representation libraries obtained through digestion with restriction enzymes--generically known as restriction site associated DNA sequencing (RAD-seq)--is a common strategy to generate genome-wide genotypic and sequence data from eukaryotes. A critical design element of any RAD-seq study is knowledge of the approximate number of genetic markers that can be obtained for a taxon using different restriction enzymes, as this number determines the scope of a project, and ultimately defines its success. This number can only be directly determined if a reference genome sequence is available, or it can be estimated if the genome size and restriction recognition sequence probabilities are known. However, both scenarios are uncommon for nonmodel species. Here, we performed systematic in silico surveys of recognition sequences, for diverse and commonly used type II restriction enzymes across the eukaryotic tree of life. Our observations reveal that recognition sequence frequencies for a given restriction enzyme are strikingly variable among broad eukaryotic taxonomic groups, being largely determined by phylogenetic relatedness. We demonstrate that genome sizes can be predicted from cleavage frequency data obtained with restriction enzymes targeting "neutral" elements. Models based on genomic compositions are also effective tools to accurately calculate probabilities of recognition sequences across taxa, and can be applied to species for which reduced representation data are available (including transcriptomes and neutral RAD-seq data sets). The analytical pipeline developed in this study, PredRAD (https://github.com/phrh/PredRAD), and the resulting databases constitute valuable resources that will help guide the design of any study using RAD-seq or related methods.
通过用限制酶消化获得的简化代表性文库的高通量测序——一般称为限制性位点相关DNA测序(RAD-seq)——是从真核生物中生成全基因组基因型和序列数据的常用策略。任何RAD-seq研究的一个关键设计要素是了解使用不同限制酶可以为一个分类群获得的遗传标记的大致数量,因为这个数量决定了一个项目的范围,并最终决定其成败。只有在有参考基因组序列的情况下才能直接确定这个数量,或者如果已知基因组大小和限制识别序列概率,也可以进行估计。然而,这两种情况对于非模式物种来说都不常见。在这里,我们对真核生物生命树中各种常用的II型限制酶的识别序列进行了系统的计算机模拟调查。我们的观察结果表明,给定限制酶的识别序列频率在广泛的真核生物分类群中存在显著差异,这在很大程度上由系统发育相关性决定。我们证明,可以根据针对“中性”元件的限制酶获得的切割频率数据来预测基因组大小。基于基因组组成的模型也是准确计算不同分类群识别序列概率的有效工具,并且可以应用于有简化代表性数据可用的物种(包括转录组和中性RAD-seq数据集)。本研究开发的分析流程PredRAD(https://github.com/phrh/PredRAD)以及由此产生的数据库构成了宝贵的资源,将有助于指导任何使用RAD-seq或相关方法的研究设计。