Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario, Canada.
PLoS One. 2012;7(4):e33971. doi: 10.1371/journal.pone.0033971. Epub 2012 Apr 6.
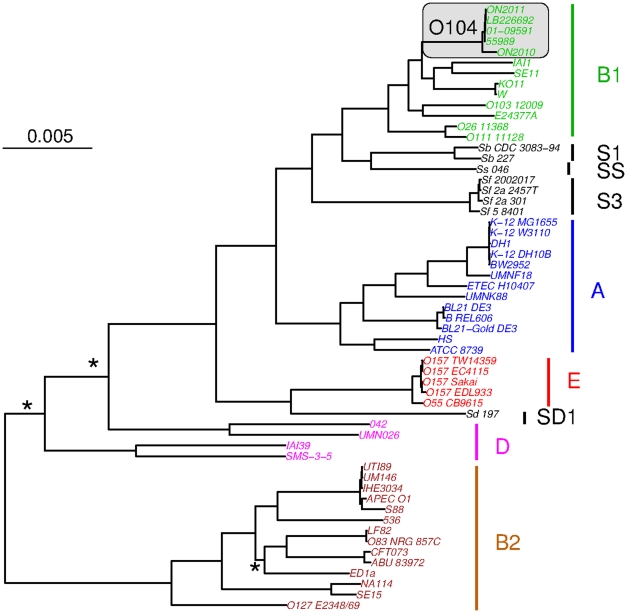
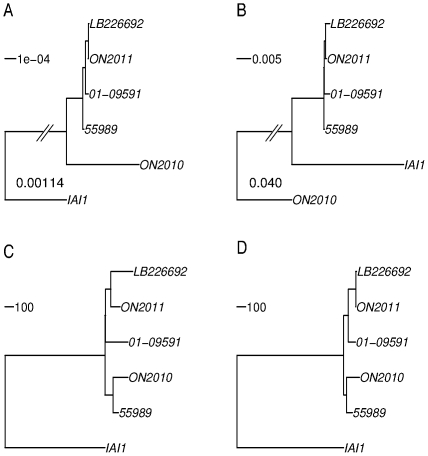
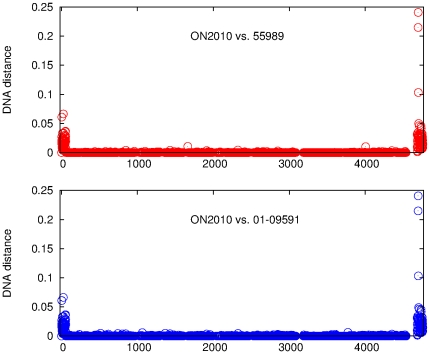
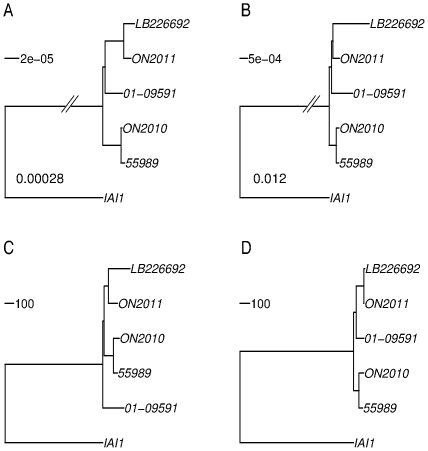
Escherichia coli O104:H4 was identified as an emerging pathogen during the spring and summer of 2011 and was responsible for a widespread outbreak that resulted in the deaths of 50 people and sickened over 4075. Traditional phenotypic and genotypic assays, such as serotyping, pulsed field gel electrophoresis (PFGE), and multilocus sequence typing (MLST), permit identification and classification of bacterial pathogens, but cannot accurately resolve relationships among genotypically similar but pathotypically different isolates. To understand the evolutionary origins of E. coli O104:H4, we sequenced two strains isolated in Ontario, Canada. One was epidemiologically linked to the 2011 outbreak, and the second, unrelated isolate, was obtained in 2010. MLST analysis indicated that both isolates are of the same sequence type (ST678), but whole-genome sequencing revealed differences in chromosomal and plasmid content. Through comprehensive phylogenetic analysis of five O104:H4 ST678 genomes, we identified 167 genes in three gene clusters that have undergone homologous recombination with distantly related E. coli strains. These recombination events have resulted in unexpectedly high sequence diversity within the same sequence type. Failure to recognize or adjust for homologous recombination can result in phylogenetic incongruence. Understanding the extent of homologous recombination among different strains of the same sequence type may explain the pathotypic differences between the ON2010 and ON2011 strains and help shed new light on the emergence of this new pathogen.
大肠杆菌 O104:H4 是 2011 年春夏期间被鉴定为新兴病原体的一种,它引发了一次广泛的爆发,导致 50 人死亡,超过 4075 人生病。传统的表型和基因型检测方法,如血清型鉴定、脉冲场凝胶电泳(PFGE)和多位点序列分型(MLST),可以识别和分类细菌病原体,但无法准确解析基因型相似但表型不同的分离株之间的关系。为了了解大肠杆菌 O104:H4 的进化起源,我们对在加拿大安大略省分离的两株菌进行了测序。其中一株与 2011 年的爆发有关,另一株则是在 2010 年获得的,与爆发无关。MLST 分析表明,两株菌均属于相同的序列型(ST678),但全基因组测序显示其染色体和质粒内容存在差异。通过对五个 O104:H4 ST678 基因组的综合系统发育分析,我们在三个基因簇中鉴定出了 167 个与远缘大肠杆菌菌株发生同源重组的基因。这些重组事件导致了同一序列型内的序列高度多样性。未能识别或调整同源重组可能导致系统发育不一致。了解同一序列型不同菌株之间同源重组的程度,可以解释 ON2010 和 ON2011 两株菌之间的表型差异,并有助于深入了解这种新病原体的出现。