Faculty of Medicine, Yamagata University, Yamagata, Japan.
PLoS Genet. 2012;8(4):e1002625. doi: 10.1371/journal.pgen.1002625. Epub 2012 Apr 5.
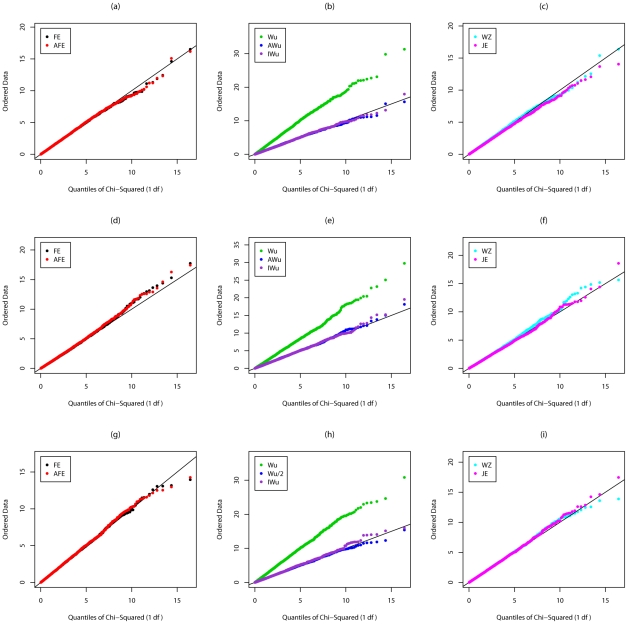
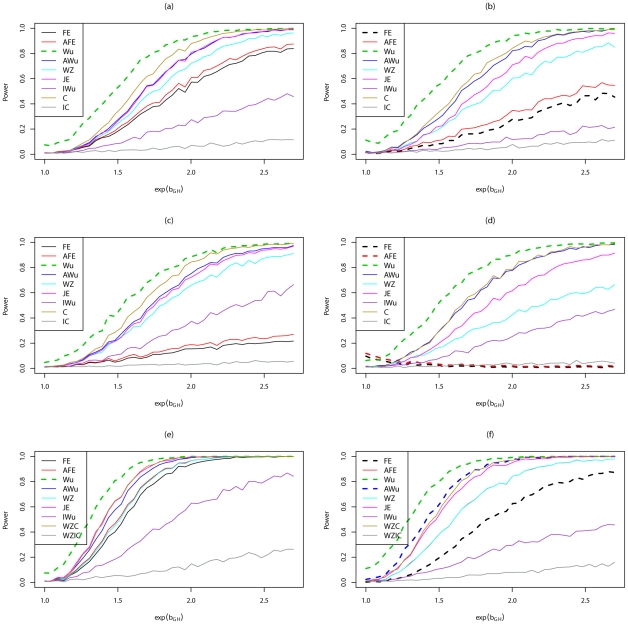
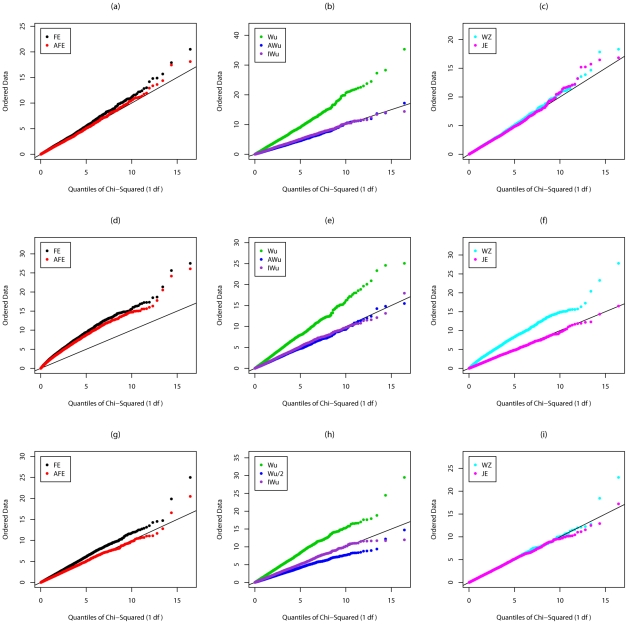
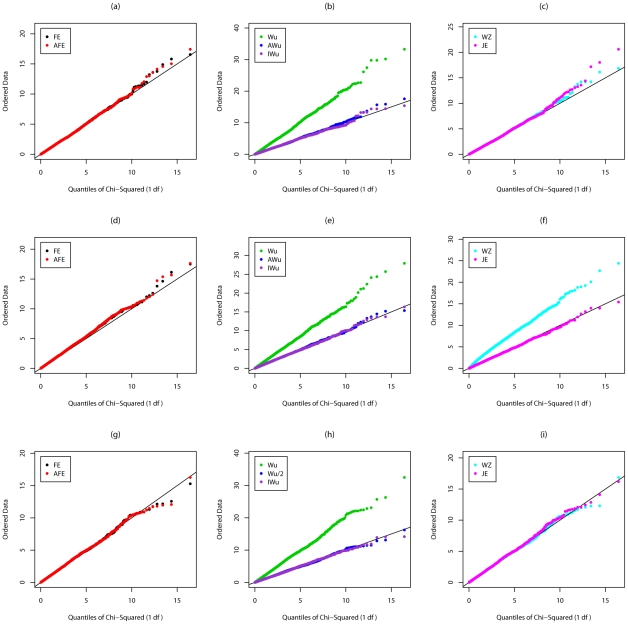
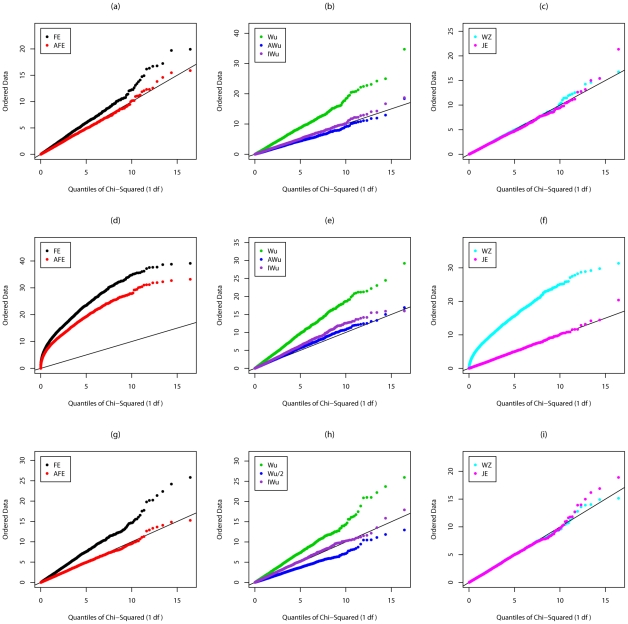
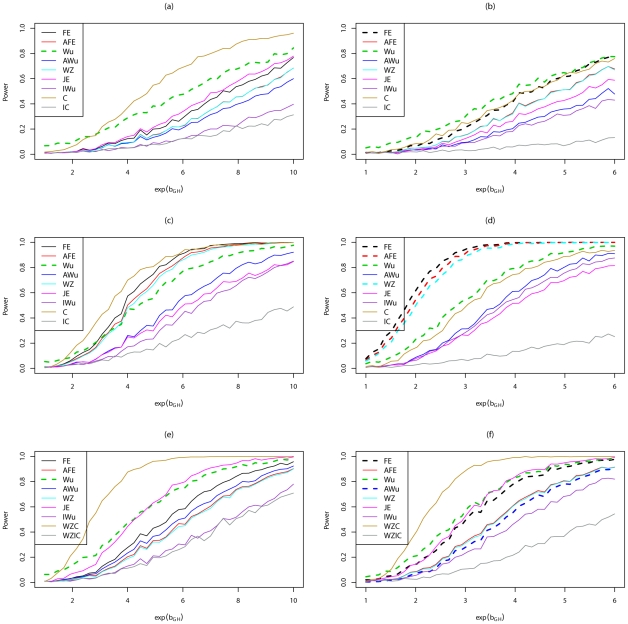
Recently, Wu and colleagues [1] proposed two novel statistics for genome-wide interaction analysis using case/control or case-only data. In computer simulations, their proposed case/control statistic outperformed competing approaches, including the fast-epistasis option in PLINK and logistic regression analysis under the correct model; however, reasons for its superior performance were not fully explored. Here we investigate the theoretical properties and performance of Wu et al.'s proposed statistics and explain why, in some circumstances, they outperform competing approaches. Unfortunately, we find minor errors in the formulae for their statistics, resulting in tests that have higher than nominal type 1 error. We also find minor errors in PLINK's fast-epistasis and case-only statistics, although theory and simulations suggest that these errors have only negligible effect on type 1 error. We propose adjusted versions of all four statistics that, both theoretically and in computer simulations, maintain correct type 1 error rates under the null hypothesis. We also investigate statistics based on correlation coefficients that maintain similar control of type 1 error. Although designed to test specifically for interaction, we show that some of these previously-proposed statistics can, in fact, be sensitive to main effects at one or both loci, particularly in the presence of linkage disequilibrium. We propose two new "joint effects" statistics that, provided the disease is rare, are sensitive only to genuine interaction effects. In computer simulations we find, in most situations considered, that highest power is achieved by analysis under the correct genetic model. Such an analysis is unachievable in practice, as we do not know this model. However, generally high power over a wide range of scenarios is exhibited by our joint effects and adjusted Wu statistics. We recommend use of these alternative or adjusted statistics and urge caution when using Wu et al.'s originally-proposed statistics, on account of the inflated error rate that can result.
最近,Wu 及其同事 [1] 提出了两种用于使用病例/对照或仅病例数据进行全基因组相互作用分析的新统计方法。在计算机模拟中,他们提出的病例/对照统计量优于竞争方法,包括 PLINK 中的快速上位效应选项和正确模型下的逻辑回归分析;然而,其优越性能的原因并未得到充分探讨。在这里,我们研究了 Wu 等人提出的统计方法的理论性质和性能,并解释了为什么在某些情况下它们优于竞争方法。不幸的是,我们发现他们的统计公式存在小错误,导致检验的Ⅰ型错误率高于名义值。我们还发现了 PLINK 的快速上位效应和仅病例统计中的小错误,尽管理论和模拟表明这些错误对Ⅰ型错误率只有微不足道的影响。我们提出了这四种统计方法的调整版本,这些调整版本在零假设下理论上和计算机模拟中都保持正确的Ⅰ型错误率。我们还研究了基于相关系数的统计方法,这些方法在保持Ⅰ型错误率控制相似的情况下也能保持类似的效果。虽然这些统计方法是专门为检验相互作用而设计的,但我们发现其中一些先前提出的统计方法实际上可能对一个或两个位点的主效应敏感,特别是在存在连锁不平衡的情况下。我们提出了两种新的“联合效应”统计方法,在疾病罕见的情况下,这些方法只对真正的相互作用效应敏感。在计算机模拟中,我们发现,在大多数考虑的情况下,在正确的遗传模型下进行分析可以获得最高的功效。然而,由于我们不知道这个模型,实际上这种分析是不可能的。然而,在广泛的场景下,我们的联合效应和调整后的 Wu 统计方法通常表现出较高的功效。我们建议使用这些替代或调整后的统计方法,并鉴于可能导致的误差率升高,谨慎使用 Wu 等人最初提出的统计方法。