Department of Pharmacology, University of Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Boulevard, Dallas, Texas 75390-9050, USA.
J Phys Chem B. 2012 Jun 21;116(24):7088-101. doi: 10.1021/jp3019759. Epub 2012 Jun 6.
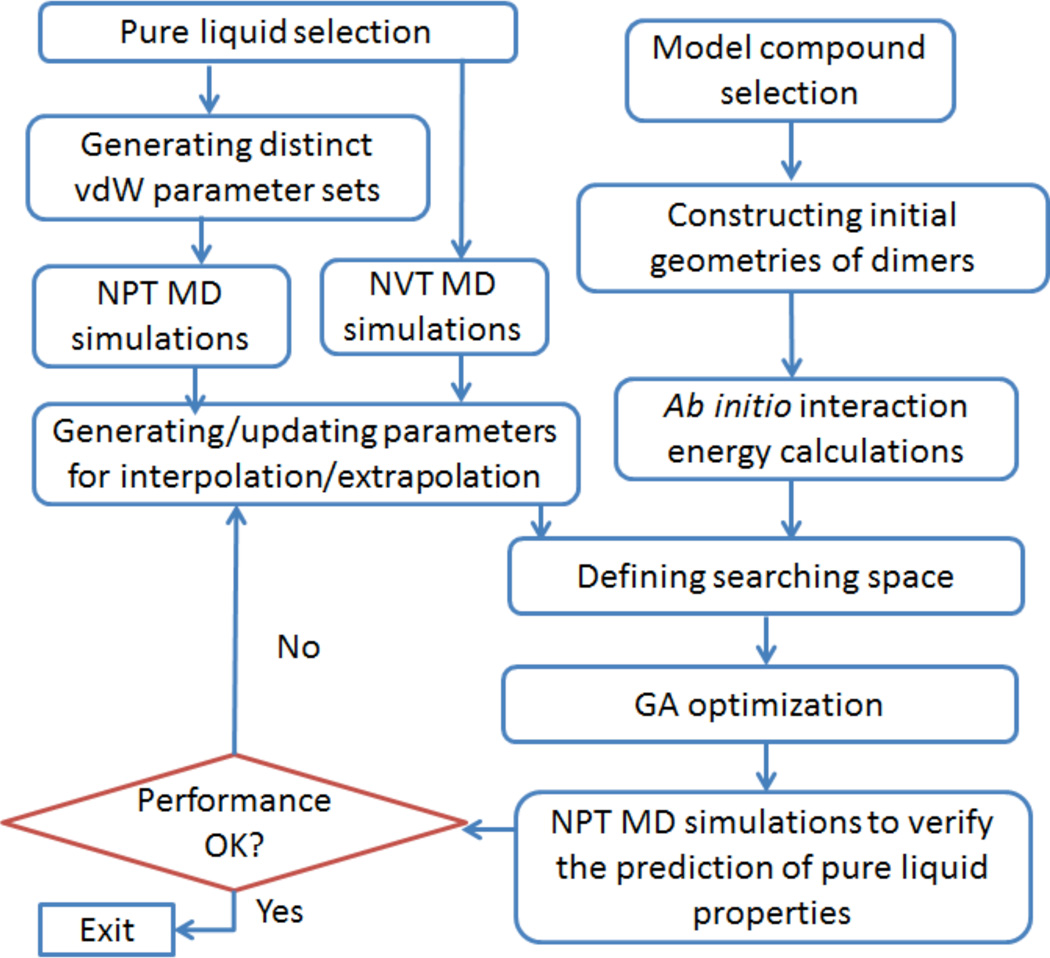
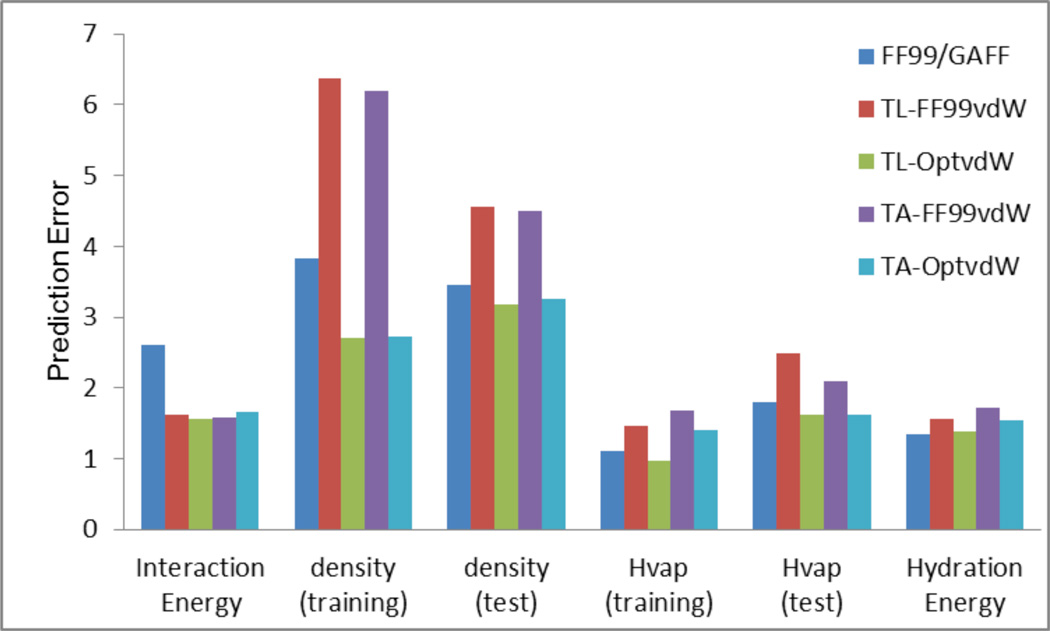
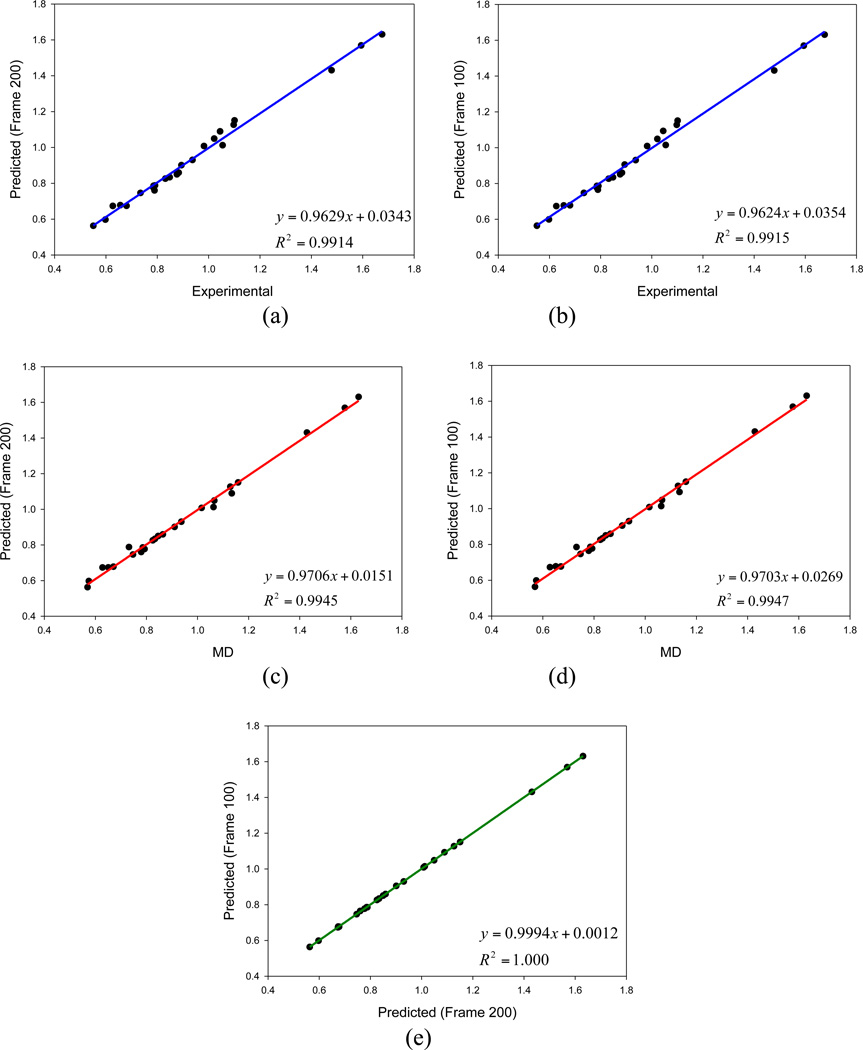
In the previous publications of this series, we presented a set of Thole induced dipole interaction models using four types of screening functions. In this work, we document our effort to refine the van der Waals parameters for the Thole polarizable models. Following the philosophy of AMBER force field development, the van der Waals (vdW) parameters were tuned for the Thole model with linear screening function to reproduce both the ab initio interaction energies and the experimental densities of pure liquids. An in-house genetic algorithm was applied to maximize the fitness of "chromosomes" which is a function of the root-mean-square errors (RMSE) of interaction energy and liquid density. To efficiently explore the vdW parameter space, a novel approach was developed to estimate the liquid densities for a given vdW parameter set using the mean residue-residue interaction energies through interpolation/extrapolation. This approach allowed the costly molecular dynamics simulations be performed at the end of each optimization cycle only and eliminated the simulations during the cycle. Test results show notable improvements over the original AMBER FF99 vdW parameter set, as indicated by the reduction in errors of the calculated pure liquid densities (d), heats of vaporization (H(vap)), and hydration energies. The average percent error (APE) of the densities of 59 pure liquids was reduced from 5.33 to 2.97%; the RMSE of H(vap) was reduced from 1.98 to 1.38 kcal/mol; the RMSE of solvation free energies of 15 compounds was reduced from 1.56 to 1.38 kcal/mol. For the interaction energies of 1639 dimers, the overall performance of the optimized vdW set is slightly better than the original FF99 vdW set (RMSE of 1.56 versus 1.63 kcal/mol). The optimized vdW parameter set was also evaluated for the exponential screening function used in the Amoeba force field to assess its applicability for different types of screening functions. Encouragingly, comparable performance was observed when the optimized vdW set was combined with the Thole Amoeba-like polarizable model, particularly for the interaction energy and liquid density calculations. Thus, the optimized vdW set is applicable to both types of Thole models with either linear or Amoeba-like screening functions.
在本系列的先前出版物中,我们提出了一组使用四种类型的屏蔽函数的 Thole 诱导偶极相互作用模型。在这项工作中,我们记录了我们努力改进 Thole 极化模型的范德华参数的工作。遵循 AMBER 力场开发的理念,我们使用线性屏蔽函数对具有范德华(vdW)参数的 Thole 模型进行了调整,以重现从头算相互作用能和纯液体的实验密度。应用内部遗传算法,使“染色体”的适合度最大化,“染色体”是相互作用能和液体密度均方根误差(RMSE)的函数。为了有效地探索 vdW 参数空间,我们开发了一种新方法,通过插值/外推通过平均残基-残基相互作用能来估算给定 vdW 参数集的液体密度。这种方法允许在每个优化周期的末尾仅执行昂贵的分子动力学模拟,并在周期内消除模拟。测试结果表明,与原始 AMBER FF99 vdW 参数集相比,有了显著的改进,这体现在计算得到的纯液体密度(d)、蒸发热(H(vap))和水合能的误差减小上。59 种纯液体密度的平均百分比误差(APE)从 5.33 降低到 2.97%;H(vap)的 RMSE 从 1.98 降低到 1.38 kcal/mol;15 种化合物的溶剂化自由能的 RMSE 从 1.56 降低到 1.38 kcal/mol。对于 1639 个二聚体的相互作用能,优化后的 vdW 集的整体性能略优于原始 FF99 vdW 集(RMSE 为 1.56 对 1.63 kcal/mol)。还评估了优化后的 vdW 参数集在用于 Amoeba 力场的指数屏蔽函数中的适用性,以评估其在不同类型的屏蔽函数中的适用性。令人鼓舞的是,当将优化后的 vdW 集与 Thole 类 amoeba 极化模型结合使用时,观察到了类似的性能,特别是在相互作用能和液体密度的计算中。因此,优化后的 vdW 集适用于具有线性或 amoeba 样屏蔽函数的两种类型的 Thole 模型。