Department of Biostatistics and Computational Biology, Dana-Farber Cancer Institute and Harvard School of Public Health, Boston, MA 02215, USA.
Nucleic Acids Res. 2012 Sep 1;40(17):e135. doi: 10.1093/nar/gks395. Epub 2012 May 29.
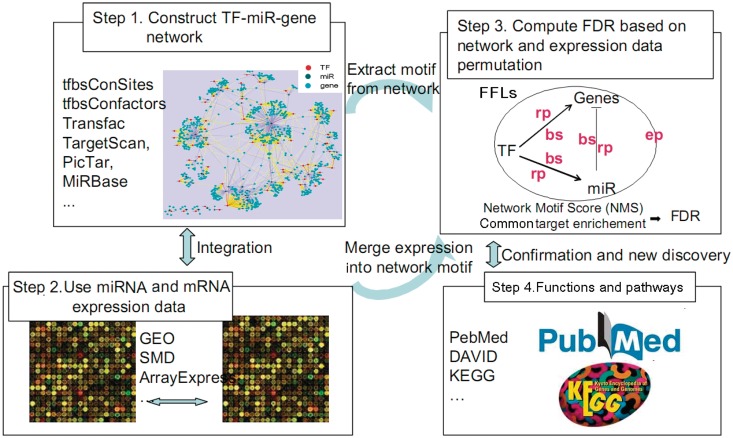
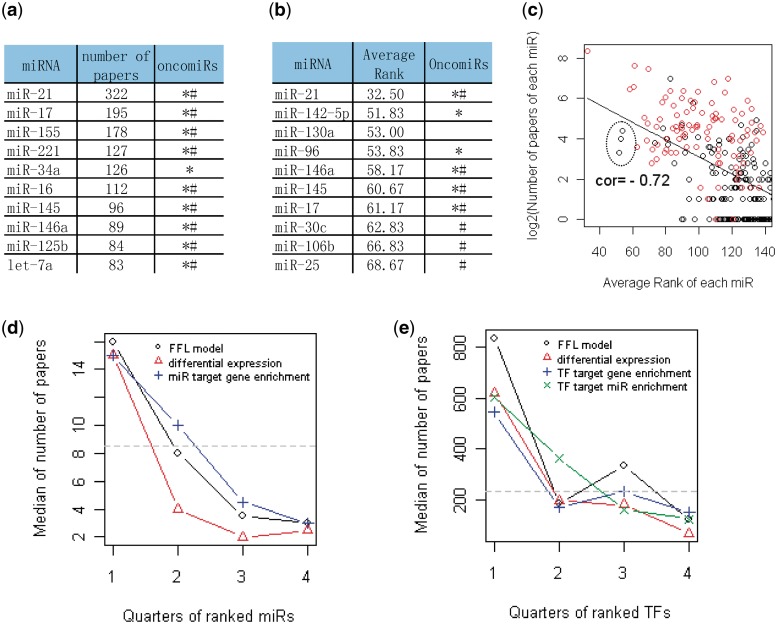
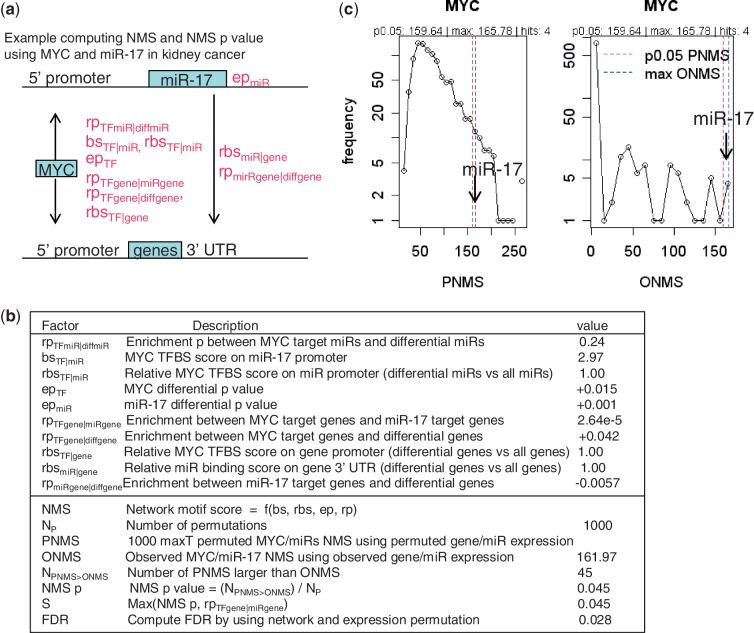
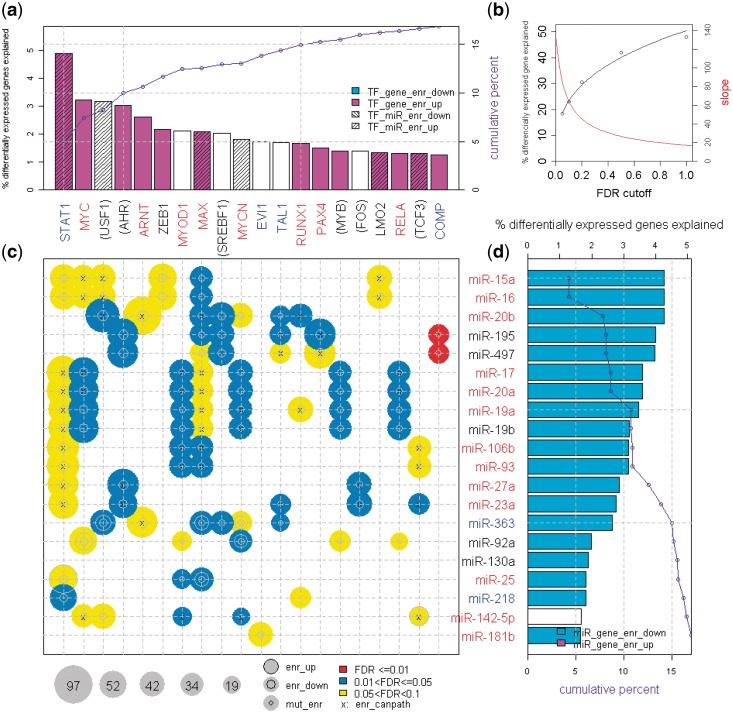
We describe here a novel method for integrating gene and miRNA expression profiles in cancer using feed-forward loops (FFLs) consisting of transcription factors (TFs), miRNAs and their common target genes. The dChip-GemiNI (Gene and miRNA Network-based Integration) method statistically ranks computationally predicted FFLs by their explanatory power to account for differential gene and miRNA expression between two biological conditions such as normal and cancer. GemiNI integrates not only gene and miRNA expression data but also computationally derived information about TF-target gene and miRNA-mRNA interactions. Literature validation shows that the integrated modeling of expression data and FFLs better identifies cancer-related TFs and miRNAs compared to existing approaches. We have utilized GemiNI for analyzing six data sets of solid cancers (liver, kidney, prostate, lung and germ cell) and found that top-ranked FFLs account for ∼20% of transcriptome changes between normal and cancer. We have identified common FFL regulators across multiple cancer types, such as known FFLs consisting of MYC and miR-15/miR-17 families, and novel FFLs consisting of ARNT, CREB1 and their miRNA partners. The results and analysis web server are available at http://www.canevolve.org/dChip-GemiNi.
我们在这里描述了一种新的方法,用于使用包含转录因子 (TF)、miRNA 及其共同靶基因的前馈回路 (FFL) 整合癌症中的基因和 miRNA 表达谱。dChip-GemiNI(基于基因和 miRNA 网络的整合)方法通过其对两种生物条件(如正常和癌症)之间的差异基因和 miRNA 表达的解释能力,对计算预测的 FFL 进行统计排序。GemiNI 不仅整合了基因和 miRNA 表达数据,还整合了关于 TF-靶基因和 miRNA-mRNA 相互作用的计算衍生信息。文献验证表明,与现有方法相比,表达数据和 FFL 的综合建模可以更好地识别与癌症相关的 TF 和 miRNA。我们已经利用 GemiNI 分析了六个实体瘤(肝、肾、前列腺、肺和生殖细胞)数据集,发现排名靠前的 FFL 约占正常和癌症之间转录组变化的 20%。我们已经确定了多个癌症类型中常见的 FFL 调节剂,例如由 MYC 和 miR-15/miR-17 家族组成的已知 FFL,以及由 ARNT、CREB1 和它们的 miRNA 伙伴组成的新型 FFL。结果和分析网络服务器可在 http://www.canevolve.org/dChip-GemiNi 上获得。